Você já ouviu falar da Síndrome de Crouzon? Também conhecida como disostose crânio-facial tipo I, é uma doença rara de origem genética, caracterizada por comprometer o desenvolvimento do esqueleto crânio-facial.

Hoje vamos abordar tudo sobre essa doença, sua causa, sintomas, diagnóstico e tratamento. Apesar de ser incomum, possui 50% de risco de transmissão quando um dos pais é portador.
Índice:
O que é a Síndrome de Crouzon?
Esta síndrome é caracterizada por anomalias crânios-faciais causadas por perda precoce de flexibilidade do crânio, presentes desde o nascimento e com tendência a agravar-se com o tempo.
Não há distinção de acometimento quanto ao sexo, porém quando as cranioestenoses são dos tipos sagitais e metópicas sua predominância aumenta no sexo masculino, enquanto a cranioestenose coronal é mais encontrada no sexo feminino.
Essa síndrome pode ser evidenciada ao nascimento ou durante a infância, sendo progressiva com o início do primeiro ano de vida ou, mais freqüentemente, aparecendo apenas aos dois anos de idade. Existem ainda formas precoces e congênitas da doença nas quais a sinostose começa ainda dentro do útero e já é manifestada ao nascimento, com deformidades faciais como a hipoplasia maxilar superior, responsável por dificuldades respiratórias, e exoftalmia.
Essa condição tem por características básicas deformidades do crânio, anomalias faciais e exoftalmia, conjunto de sintomas que hoje constituem a chamada tríade da doença de Crouzon. Nesta, o fechamento prematuro de suturas cranianas e a sinostose prematura de suturas centro-faciais e da base do crânio, conferem-lhe uma configuração braquiocefálica, ou seja, crânio alto e em forma de torre.
Uma vez que a sutura se torne fundida, o crescimento perpendicular a esta se torna restrito e os ossos fundidos agem como uma única estrutura óssea, ocorrendo, assim, crescimento compensatório nas suturas abertas restantes para dar continuidade ao desenvolvimento do cérebro ocasionando crescimento ósseo anormal e produção de deformidades faciais.
Além disso, tal situação provoca um desequilíbrio entre o crescimento ósseo (craniano) e de partes moles (encefálico), levando por vezes ao surgimento de hipertensão intracraniana (HIC) pela restrição do crescimento cerebral, que pode acarretar em prejuízo no desenvolvimento cognitivo destes pacientes. Em síntese, é uma mutação de um gene, chamado FGFR2, responsável por coordenar o encaixe dos ossos do crânio e rosto.
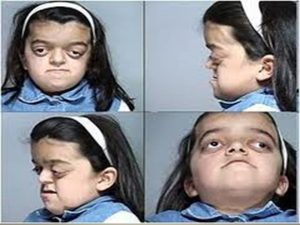
Seus sinais exteriores são deformidades na cabeça, ossos faciais afundados, olhos separados, testa protuberante. Através de cirurgias é possível a separação e reconstrução dos ossos do crânio, realinhamento do maxilar, olhos e nariz. (fonte: Revista Veja, edição 2293, nº 44, 31 de outubro de 2012).
Quais os sintomas da Síndrome de Crouzon?
Entre as dificuldades afetadas por um paciente com essa síndrome, destacam-se problemas mentais, em que o QI é muito abaixo do normal, problemas visuais, problemas auditivos, respiratórios, digestórios (relacionados à mastigação), cardíacos, morfológicos, anatômicos e de comunicação (relacionados à fala). Além disso, os portadores dessa doença sofrem com o déficit de atenção, dificuldade de aprendizado e alteração comportamental.
Os principais sinais clínicos são craniossinostose, hipertelorismo, exoftalmia, estrabismo externo, “nariz de bico de papagaio”, lábio superior curto, hipoplasticidade maxilar e relativo prognatismo mandibular, determinando um aspecto de hipoplasia centrofacial. É uma afecção hereditária com transmissão autossômica dominante com 100% de penetrância e larga escala da expressão fenotípica.
Sintomas relacionados com a visão
Órbitas rasas, exoftalmia, proptose ocular bilateral (protrusão anormal do globo ocular), hipertelorismo (deformação congênita do crânio e da face, manifestando-se por afastamento excessivo dos olhos, com alargamento da raiz do nariz), estrabismo divergente (o desvio de um dos olhos é para dentro, voltado para o nariz), atrofia óptica (perda de algumas ou da maioria das fibras do nervo óptico), conjuntivite ou ceratoconjuntivite de exposição e redução da acuidade visual (característica do olho de reconhecer dois pontos muito próximos).
Em raros casos podem estar presentes nistagmo (movimentos oculares oscilatórios, rítmicos e repetitivos dos olhos), anisocoria (tamanho desigual das pupilas), catarata, glaucoma e luxação (deslocamento de um ou mais ossos de uma articulação) do globo ocular.
Sintomas relacionados com a audição
Hipoacusia (surdez), malformações do ouvido médio, perda auditiva condutiva não progressiva em um terço dos casos, e ainda perda auditiva mista.
Sintomas relacionados com a respiração
Angústia respiratória aguda, dispneia (falta de ar) do tipo polipneia (respiração muito rápida e ofegante) e apneia do sono (interrupção da entrada de ar nasal e oral), seios paranasais reduzidos.
Sintomas cardíacos
Malformações ventriculares.
Sintomas relacionados a características morfológicas e dermatológicas
Fronte larga, nariz com aspecto adunco, Acantose Nigricans (desordem que causa manchas aveludadas, cor marrom a preto, geralmente no pescoço, por baixo do braço, ou na região inguinal), sendo a principal manifestação dermatológica da síndrome de Crouzon, sendo detectável após a infância.
Sintomas relacionados à anatomia
Achatamento da região occipital (região posterior do crânio conhecido como nuca), relativa protuberância fronto-occipital, hipoplasia centrofacial, moderada braquicefalia (sutura coronal se funde prematuramente), impressões cerebriformes, alargamento da fossa hipofisária.
Anomalias dentárias em portadores da síndrome de Crouzon
Como vimos até agora, a síndrome de Crouzon envolve diversos sintomas que são característicos dessa anomalia. No entanto, os pacientes portadores dessa síndrome apresentam certa prevalência de anomalias dentárias, principalmente relacionadas à erupção, provavelmente resultante da grande hipoplasia maxilar apresentada.
Em geral, os pacientes com a síndrome de Crouzon apresentam hipoplasia centrofacial e maxilar e prognatismo mandibular (mandíbula extremamente pronunciada), e em alguns casos podem apresentar ainda a atresia maxilar, que é uma relação anormal entre os maxilares, onde os dentes superiores estão dispostos de forma mais estreita que os inferiores, levando, assim, a uma má oclusão dentária.
Alguns indivíduos podem apresentar arco dental maxilar em forma de V com dentes muito espaçados e aglomerados. Na boca, existem casos de pacientes com estreitamento do palato duro, fissura congênita do céu da boca, a fenda palatina, palato em ogiva, com uma região côncava no osso palatal, e úvula bífida.
Além disso, existem ainda casos de apinhamento dentário, problema geralmente relacionado a um excesso de volume dentário ou a uma base óssea pequena demais para comportar os dentes.
Indivíduos com a síndrome de Crouzon, além destas características, podem ainda apresentar alterações na forma dos dentes, como a macrodontia, dentes com tamanho anatomicamente maior que o habitual, alterações no número de dentes, com anodontia ou dentes supranumerários, alterações de erupção, com dente retido, erupção retardada ou erupção ectópica. Nesses casos de problemas com a erupção, os dentes mais acometidos são os caninos superiores.
Qual a causa dessa síndrome?
Como vimos até agora, a Síndrome de Crouzon é uma condição genética rara. Ela soma aproximadamente 4% dos casos totais de craniossinostoses. Apesar de ser pouco frequente, se um dos pais possui o gene mutante, existe uma chance de 50% de transmissão para o bebê.
Mutações no gene FGFR2 causam a síndrome de Crouzon. Este gene fornece instruções para a produção de uma proteína chamada receptor do fator de crescimento de fibroblastos 2.
Entre suas múltiplas funções, esta proteína sinaliza às células imaturas como células ósseas durante o desenvolvimento embrionário. Mutações no gene FGFR2 provavelmente super estimulam a sinalização pela proteína FGFR2, que faz com que os ossos do crânio se fundam prematuramente. Também entendemos que ela pode acometer tanto o sexo feminino, como o masculino (hereditariedade autossômica dominante).
Alguns estudos consideram também que a síndrome esteja ligada à idade paterna avançada no momento da concepção.
Embora muitas das malformações físicas associadas a Apert não se apresentem em um paciente de Crouzon, acredita-se que ambas as condições tenham origens genéticas semelhantes. Na síndrome de Crouzon, a deformidade do crânio apresenta a mesma aparência da síndrome de Apert; no entanto, com Crouzon, não há fusão dos dedos das mãos e dos pés.
Diagnóstico
Nos casos de Síndrome de Crouzon, o diagnóstico precoce é essencial para proporcionar o melhor desenvolvimento possível para a criança. Por isso, pais que possuem algum caso da condição na família devem redobrar a atenção. O mapeamento genético é uma opção para quem tem histórico da síndrome.
Em alguns casos, é possível observar anomalias já em ultrassonografias realizadas no pré-natal, durante a gravidez. Nessas situações, pode ser realizada a ressonância magnética fetal, para observar assimetrias cranianas ou faciais.
O diagnóstico também costuma ser feito após o nascimento, geralmente entre o primeiro e segundo ano de vida da criança, época em que os sintomas ficam perceptíveis. As radiografias cranianas são recomendadas para avaliação das deformidades, além de tomografias computadorizadas com janela óssea.
Caso os pais, parentes e pediatra identifiquem as características da Síndrome de Crouzon, a criança deve ser encaminhada para neurocirurgião pediátrico que acompanhará o diagnóstico e o tratamento do paciente.
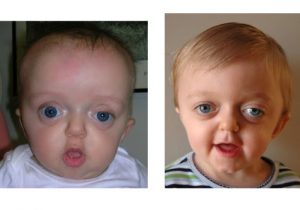
Diferença entre a síndrome de Crouzon e estenose crânio-facial
A estenose crânio-facial é uma má formação óssea (“malformação” no termo médico) decorrente da ausência ou do fechamento prematuro das suturas (pontos em que os ossos se unem) cranianas e faciais acompanhado de hipoplasia maxilar orbital (desenvolvimento precário da maxila e do globo ocular, que é responsável pelos olhos saltados).
Pode ocorrer isoladamente (como é o caso da filha do empresário Roberto Justus com a apresentadora Ticiane Pinheiro, Rafaella Justus) ou associada a mais de 70 tipos de síndromes, sendo as mais comuns as de Crouzon e Apert.
A prevalência do problema sobre a população ainda não é bem estabelecida, mas estima-se que a estenose crânio-facial acometa uma em cada 2 mil crianças no mundo. O sexo masculino é o mais afetado, com incidência três vezes maior do que em mulheres.
Quais as causas?
Como é uma má-formação de caráter genético, as causas são indeterminadas.
Diagnóstico de Estenose crânio-facial
A anormalidade pode ser descoberta por meio de um estudo radiológico, radiografias ou tomografias do crânio com reconstrução tridimensional. Exames de ressonância magnética ainda podem mostrar sinais de atrofia cerebral ou outras anormalidades.
O mapeamento ósseo também pode ser usado para confirmar o diagnóstico, assim como a investigação genética, em alguns casos. Entretanto, uma vez descoberto o problema, nada pode ser feito para evitá-lo.
Tratamento de Estenose crânio-facial
Quando a estenose crânio-facial tem impacto apenas na parte estética, paciente e médico devem discutir a necessidade de uma intervenção cirúrgica precoce para o bem-estar da criança. Nos casos em que o fechamento das suturas coloca em risco a vida ou o desenvolvimento da criança, o procedimento cirúrgico é fundamental e deve ser realizado o quanto antes.
Complicações possíveis
A não intervenção cirúrgica pode levar a deformidades permanentes da caixa craniana e das estruturas faciais associadas, que incluem deficiências estéticas, funcionais e psicossociais. Algumas delas são:
- Microcefalia: quando o cérebro atrofia porque não tem espaço para se desenvolver
- Hidrocefalia: dilatação dos ventrículos e de cavidades dentro do cérebro pelo acúmulo de líquor, líquido que hidrata e protege o órgão
- Proptose ocular: deslocamento do globo ocular.
Qual o prognóstico?
Alguns casos exigem acompanhamento fisioterápico, fonoaudiológico e ortodôntico. Se não estiver vinculado a nenhuma síndrome, o problema é apenas estético, não afetando o desenvolvimento neurológico e intelectual do indivíduo.
Como é feito o tratamento?
Não existe um tratamento específico que cure a síndrome de Crouzon, e por isso o tratamento da criança envolve a realização de cirurgias para amenizar as alterações ósseas, diminuir as pressões na cabeça e prevenir alterações do desenvolvimento da forma do crânio e tamanho do cérebro, tendo tanto efeitos estéticos como efeitos que visam melhorar o aprendizado e funcionalidade.
Idealmente, a cirurgia deve ser realizada antes de 1 ano de vida da criança, já que os ossos são mais maleáveis e mais fáceis de ajustar. Além disso, o preenchimento de falhas ósseas com próteses de metilmetacrilato tem sido utilizado na cirurgia estética para suavizar e harmonizar o contorno facial.
Além disso a criança deve fazer fisioterapia e terapia ocupacional por algum tempo. O objetivo da fisioterapia será melhorar a qualidade de vida da criança e levá-la a um desenvolvimento psicomotor o mais próximo do normal possível. Psicoterapia e fonoaudiologia também são formas complementares de tratamento, e a cirurgia plástica também é benéfica para melhorar o aspecto facial e melhorar a auto-estima do paciente.
Dados sobre a doença
A Síndrome de Crouzon é responsável por aproximadamente 4.8% de todos os casos de craniossinostose, sendo a síndrome mais comum do grupo de mais de 100 tipos de craniossinostoses.
Como dito anteriormente, a tríade composta por deformidades do crânio, anomalias faciais e exoftalmia, descrita por Crouzon em 1912, hoje forma a síndrome de Crouzon. Nesta, o fechamento prematuro de suturas cranianas e a sinostose prematura de suturas centro-faciais e da base do crânio, conferem-lhe uma configuração braquiocefálica.
Uma vez que a sutura se torne fundida, o crescimento perpendicular a esta se torna restrito e os ossos fundidos agem como uma única estrutura óssea. Ocorrem crescimento compensatório nas suturas abertas restantes para dar continuidade ao desenvolvimento do cérebro ocasionando crescimento ósseo anormal e produção de deformidades faciais.
Esta síndrome é progressiva, de início no primeiro ano de vida e aparecendo com freqüência só aos dois anos de idade. Existem ainda formas precoces congênitas nas quais a sinostose começa ainda dentro do útero e já é manifestada ao nascimento com deformidades faciais como a hipoplasia maxilar superior, responsável por dificuldades respiratórias e exoftalmia.
A raridade da Síndrome de Crouzon e sua heterogeneidade de espectros demonstram o caráter multifatorial desta patologia. Por ser caracterizada por anomalias crânios-faciais e tratar-se de uma síndrome disgenética multifatorial, faz-se necessário o aconselhamento genético e o estudo detalhado em cada indivíduo acometido por esta síndrome. Portanto, deve ser contínuo o estudo a fim de se promover o avanço no diagnóstico necessário a uma precoce abordagem multidisciplinar e início do tratamento específico, prevenindo desta forma os efeitos do diagnóstico tardio.
Se você gostou das informações ou ainda tem alguma dúvida, comente abaixo que o Cliquefarma terá o maior prazer em interagir com você!





