para o que é indicado e para que serve?
Para que serve Leucemia Linfocítica Crônica (LLC): Gazyva está indicado, juntamente com outro medicamento chamado clorambucil, para tratar pacientes adultos portadores de leucemia linfocítica crônica, que não tenham recebido outro medicamento anteriormente e que apresentem comorbidades (ocorrência simultânea de dois ou mais problemas de saúde em um mesmo indivíduo), tornando-os não elegíveis ao tratamento baseado na dose plena do medicamento fludarabina.Continue lendo...
ofertas de
Gazyva - 1000 Mg Solução ...
ofertas de Gazyva - 1000 Mg Solução ...
R$ 32.888,00
R$ 33.892,36
R$ 33.902,86
R$ 34.890,00
ATENÇÃO: O texto abaixo deve ser utilizado apenas como uma referência secundária. É um registro histórico da bula, rótulo ou manual do produto. Este texto não pode substituir a leitura das informações que acompanha o produto, cujo fabricante podem mudar a formulação, recomendação, modo de uso e alertas legais sem que sejamos previamente comunicados. Apenas as informações contidas na própria bula, rótulo ou manual que acompanha o produto é que devem estar atualizadas de acordo com a versão comercializada porém, no caso de qualquer dúvida, consulte o serviço de atendimento ao consumidor do produto ou nossa equipe.
Para que serve
Leucemia Linfocítica Crônica (LLC):
Gazyva está indicado, juntamente com outro medicamento chamado clorambucil, para tratar pacientes adultos portadores de leucemia linfocítica crônica, que não tenham recebido outro medicamento anteriormente e que apresentem comorbidades (ocorrência simultânea de dois ou mais problemas de saúde em um mesmo indivíduo), tornando-os não elegíveis ao tratamento baseado na dose plena do medicamento fludarabina.
Linfoma Folicular:
Gazyva está indicado, juntamente com outro medicamento chamado bendamustina e depois tomado sozinho, em terapia de manutenção, para tratamento de portadores de linfoma folicular que não tenham respondido ou que tenham recaído durante ou depois do tratamento com rituximabe ou com um esquema de tratamento contendo rituximabe.
Como Gazyva funciona?
Gazyva é um anticorpo do tipo IgG1 fabricado por glicoengenharia. Ele atinge um antígeno que fica na superfície de algumas células brancas do sangue chamadas de linfócitos que podem ser não malignas ou malignas, mas não atinge as células-tronco do sistema formador de células do sangue nem outras células de tecidos normais. Por serem produzidos por glicoengenharia, esses anticorpos apresentam uma atividade mais potente.
Em estudos não clínicos, isto é, que não foram realizados em seres humanos, Gazyva provocou a morte direta das células e colaborou na toxicidade celular e fagocitose (englobamento de células pelo sistema de defesa) que dependem de anticorpos recrutando células imunoefetoras (células com atividade imunológica). Também atuou na toxicidade que depende de complemento. Isso se traduz em uma redução maior das células do tipo B e maior eficácia antitumoral em animais.
Em um estudo clínico realizado com pacientes tratados com Gazyva, houve redução de células B (número de células B CD19+ < 0,07 x 109/L) no final do período de tratamento e o número continuou baixo durante o período de recuperação dos primeiros 6 meses. A recuperação de células B foi observada entre 12 a 18 meses de acompanhamento em 35% (14 de 40) pacientes sem doença progressiva e 13% (5 de 40) com doença progressiva.
Contraindicação
Você não deve utilizar este medicamento se souber que tem alergia ao obinutuzumabe ou a qualquer um dos outros componentes de Gazyva.
Como usar
Gazyva deve ser administrado por infusão intravenosa por meio de um acesso venoso exclusivo, em um ambiente onde meios de reanimação estejam imediatamente disponíveis e sob a supervisão rigorosa de um médico experiente. Infusões de Gazyva não devem ser administradas em injeção direta ou em bolus. Deve-se utilizar solução isotônica de cloreto de sódio 0,9% (soro fisiológico) como veículo de infusão.
Pré-medicação, consistindo de analgésico/antipirético, anti-histamínico e glicocorticoide, deverá ser sempre administrada antes de cada infusão de Gazyva.
O profissional da saúde saberá como preparar o medicamento.
Leucemia linfocítica crônica - Dose de Gazyva em combinação com clorambucil
A dose recomendada de Gazyva é de 1000 mg administrados nos Dias 1-2 (100 + 900 mg), 8 e 15 do primeiro ciclo de tratamento de 28 dias, seguido por 1000 mg administrados apenas no Dia 1 para cada ciclo de tratamento subsequente (Ciclos 2 a 6).
Linfoma Folicular
A dose recomendada de Gazyva é de 1000 mg administrados no Dia 1, Dia 8 e dia 15 do primeiro ciclo de tratamento de 28 dias, seguidos por 1000 mg administrados apenas no Dia 1 de cada ciclo de tratamento de 28 dias subsequente (Ciclos 2 a 6).
Pacientes que responderem ao tratamento (resposta completa ou resposta parcial) ou apresentem doença estável devem continuar a receber Gazyva 1000 mg isoladamente como terapia de manutenção uma vez a cada 2 meses até a progressão da doença ou por até dois anos.
Duração do Tratamento
Seis ciclos de tratamento, com duração de 28 dias cada.
Modificações de dose durante o tratamento
Não são recomendadas reduções de dose de Gazyva.
Ajustes de dose para populações especiais
O que devo fazer quando eu me esquecer de usar Gazyva?
Caso uma dose programada de Gazyva seja perdida, a mesma deve ser administrada assim que possível. Não omita e nem espere até a próxima dose planejada.
Seu médico saberá quando deverá ser aplicada a próxima dose de Gazyva.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Precauções
A substituição de Gazyva por qualquer outro medicamento biológico exige o consentimento do médico prescritor.
Gerais
Infusões de Gazyva não devem ser administradas em injeção direta ou em bolus.
Reações Adversas
Leucemia Linfocítica Crônica
As reações que este medicamento pode provocar foram identificadas durante o tratamento e acompanhamento em estudos clínicos, em que Gazyva foi administrado juntamente com clorambucil e a comparação foi feita com o uso de clorambucil sem Gazyva (Estágio 1) ou rituximabe mais clorambucil (Estágio 2). Oitenta e um por cento (81%) dos pacientes tratados com Gazyva com clorambucil receberam os 6 ciclos completos de tratamento, em comparação com 89% dos pacientes no braço do estudo que receberam rituximabe mais clorambucil e 67% dos pacientes no braço do estudo que receberam apenas clorambucil.
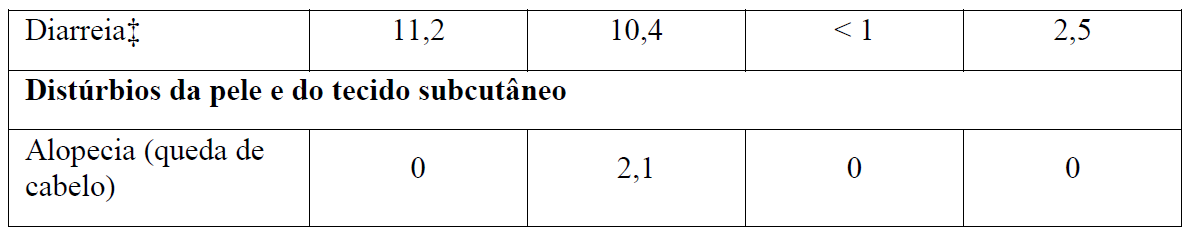
As Tabelas 1 e 2 resumem as reações adversas que ocorreram com maior frequência (diferença ? 2%) em pacientes que receberam Gazyva com clorambucil em comparação com clorambucil apenas ou rituximabe mais clorambucil, respectivamente.
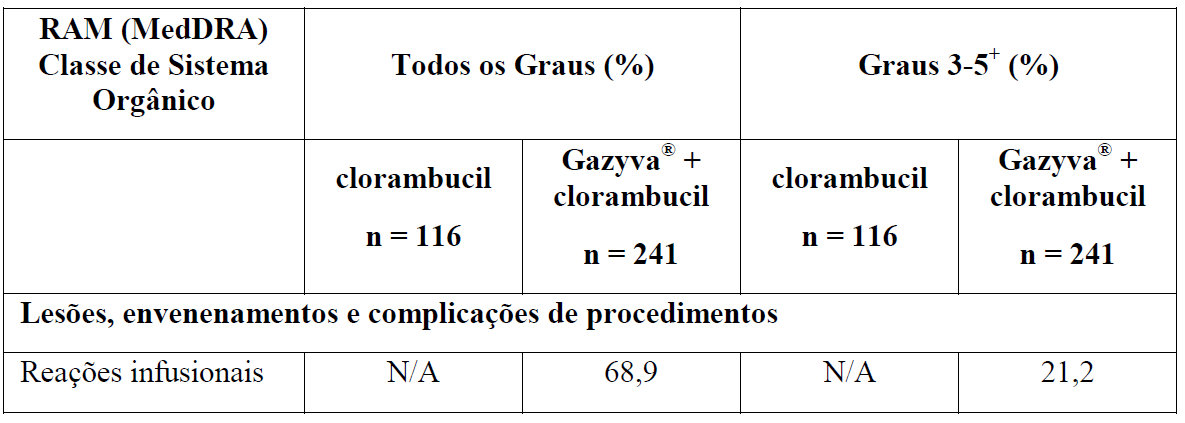
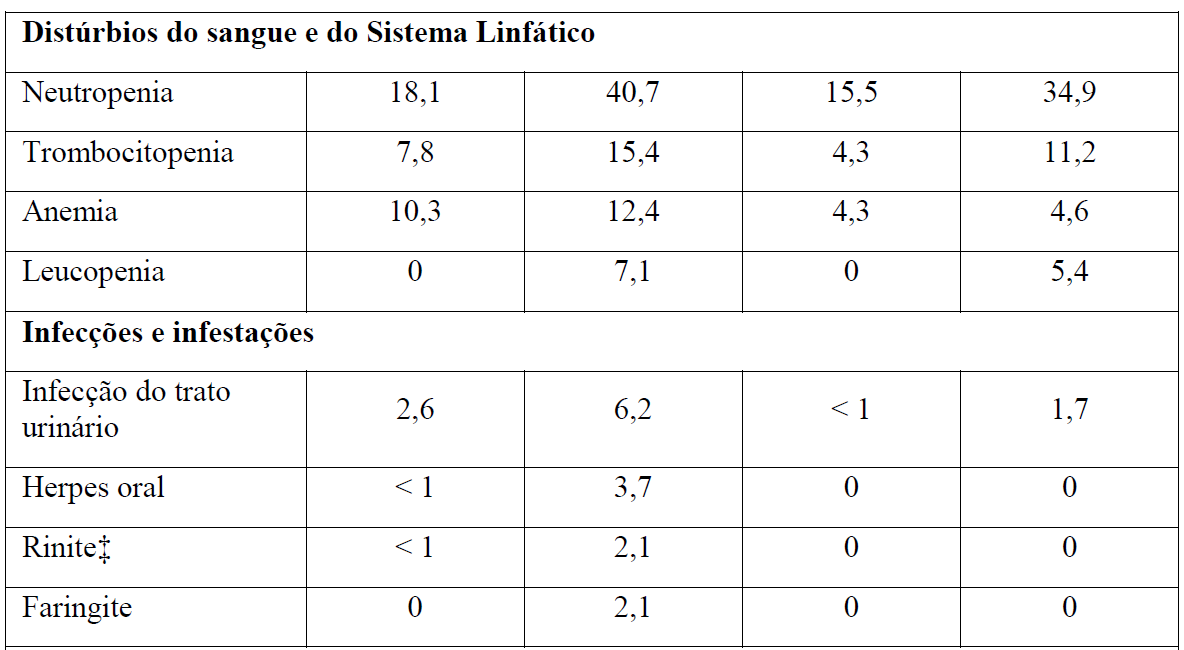
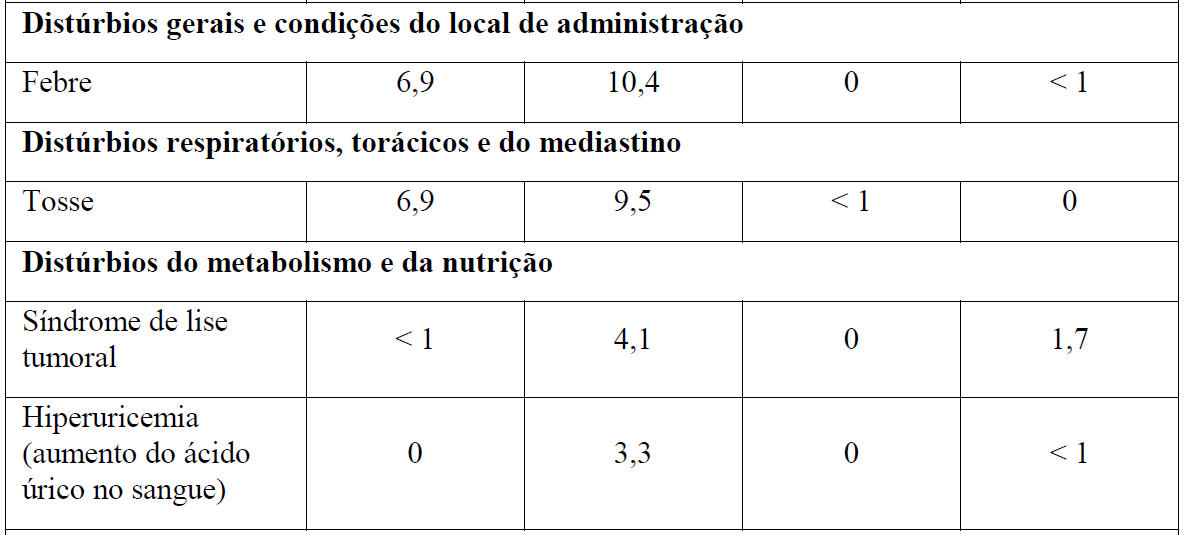
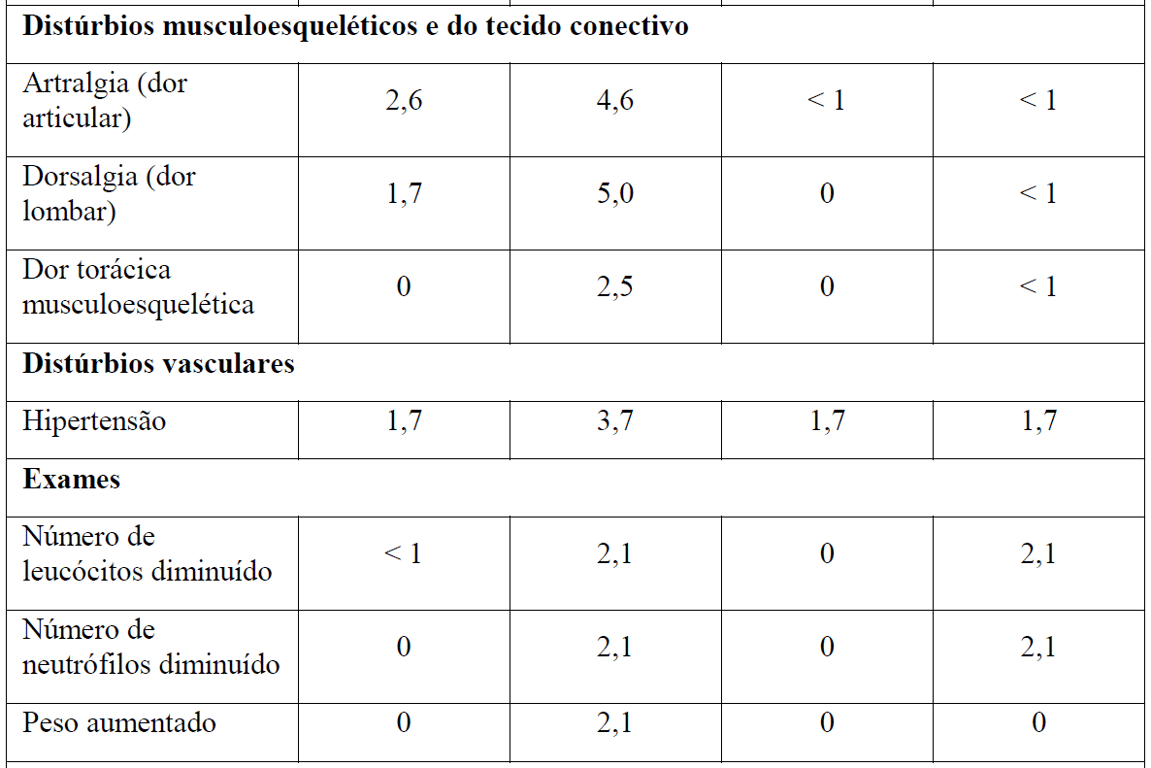

*Em todos os graus ou Graus 3-5.
+ Nenhuma reação adversa Grau 5 foi observada com uma diferença ? 2% entre os braços de tratamento.
‡ Com a atualização dos dados do Estágio 1 e Estágio 2, esse evento não foi mais reportado com uma diferença de ? 2% entre os braços de tratamento.
Tabela 2 Reações adversas reportadas com maior incidência (diferença ? 2%) em pacientes recebendo Gazyva mais clorambucil em comparação com rituximabe mais clorambucil (Estágio 2)
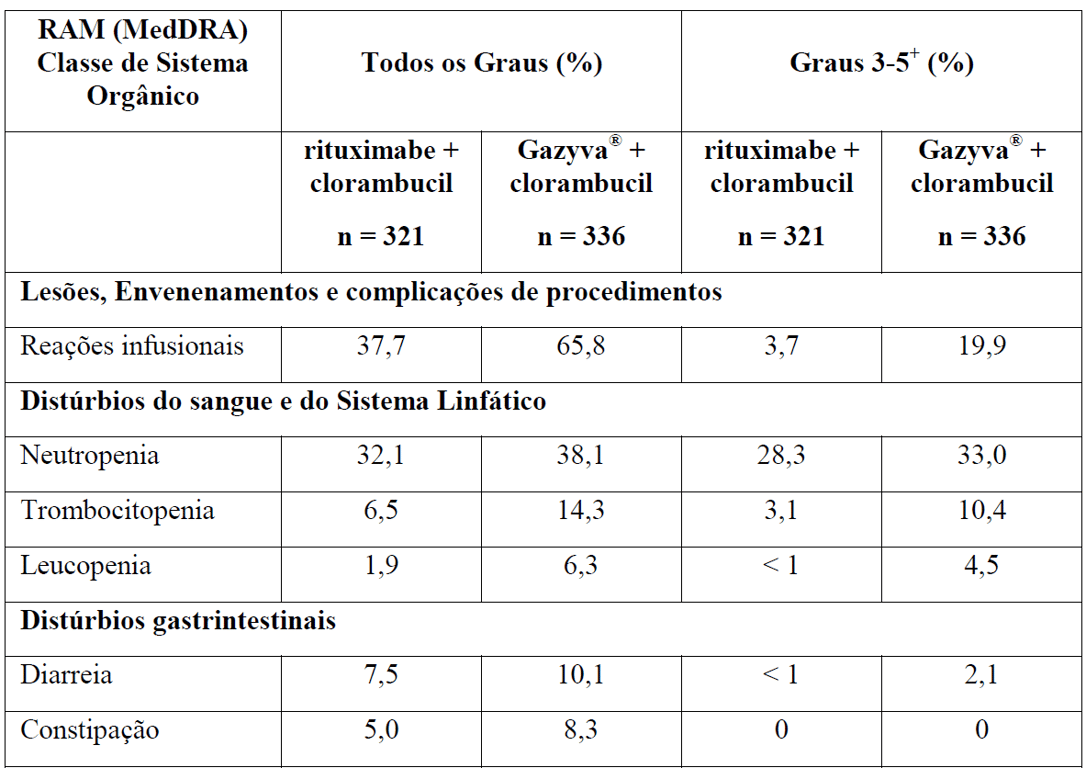
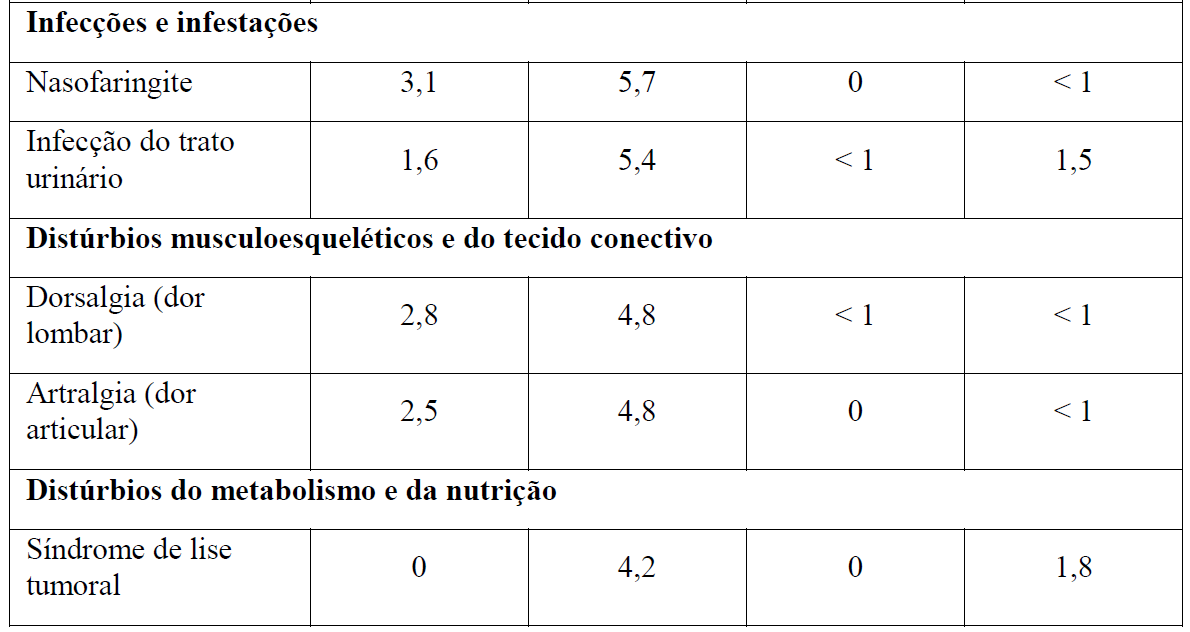
* Em todos os graus ou Graus 3-5.
+ Nenhuma reação adversa Grau 5 foi observada com uma diferença ? 2% entre os braços de tratamento.
Linfoma Não-Hodgkin
No subgrupo de pacientes com linfoma folicular, o perfil das reações adversas foi compatível com a população total de pacientes com linfoma Não-Hodgkin (LNH).
As reações adversas à medicamentos (RAMs) descritas nesta seção (baseadas em uma população de segurança de 392 pacientes com LNH indolente, dos quais 81% tinham LF) foram identificadas durante a indução, a manutenção e o acompanhamento do estudo clínico, em que Gazyva foi administrado em combinação com bendamustina (G+B) durante a indução e em monoterapia na manutenção e comparadas com bendamustina durante a indução apenas (B).
Em pacientes tratados com G+B, 79,4% receberam os 6 ciclos de tratamento com Gazyva e 75,6% receberam os 6 ciclos de B em comparação com 66,7% dos pacientes no braço B.
A Tabela 3 resume as RAMs que ocorreram com maior incidência (diferença > 2%) em pacientes que receberam G+B durante a indução seguidos por manutenção com Gazyva em comparação com B durante a indução apenas.
Tabela 3 Reações Adversas reportadas com maior incidência (diferença > 2%) em pacientes recebendo Gazyva mais bendamustina (indução) seguidos por manutenção com Gazyva em relação a bendamustina (indução) apenas
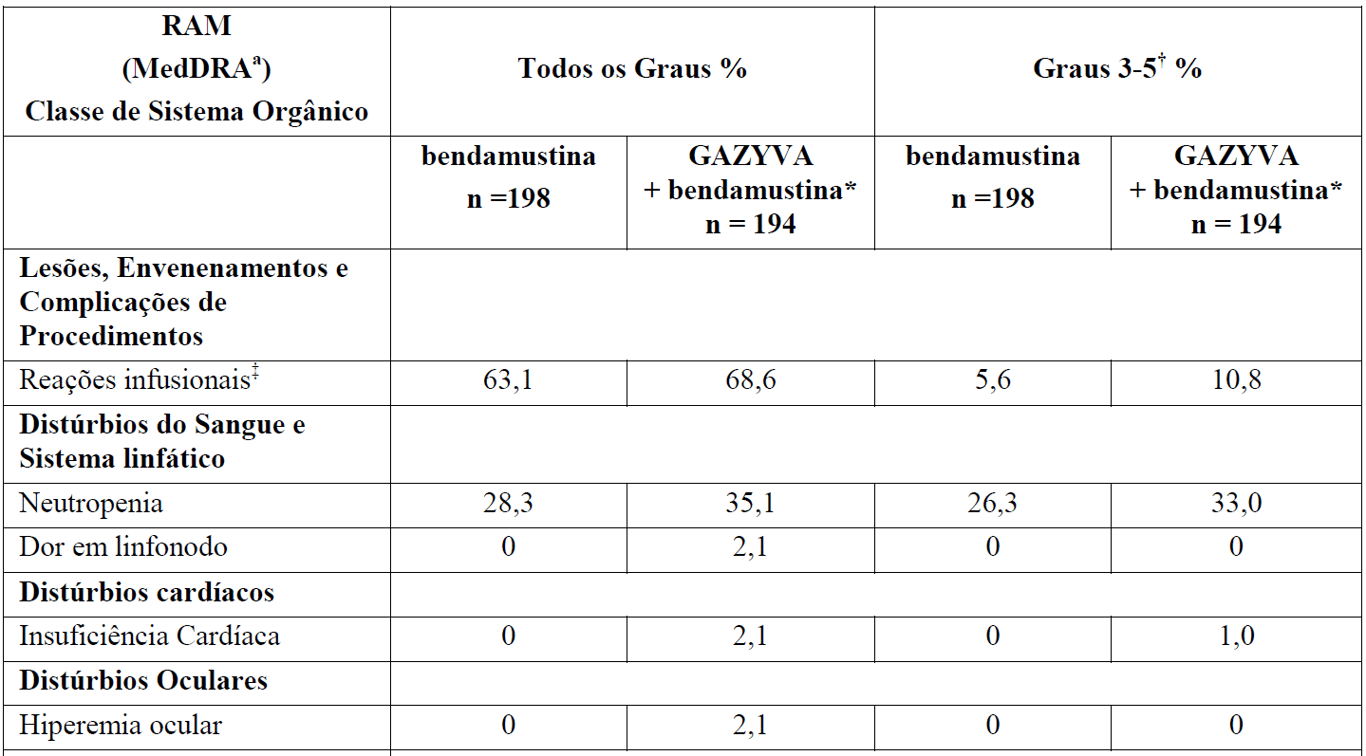
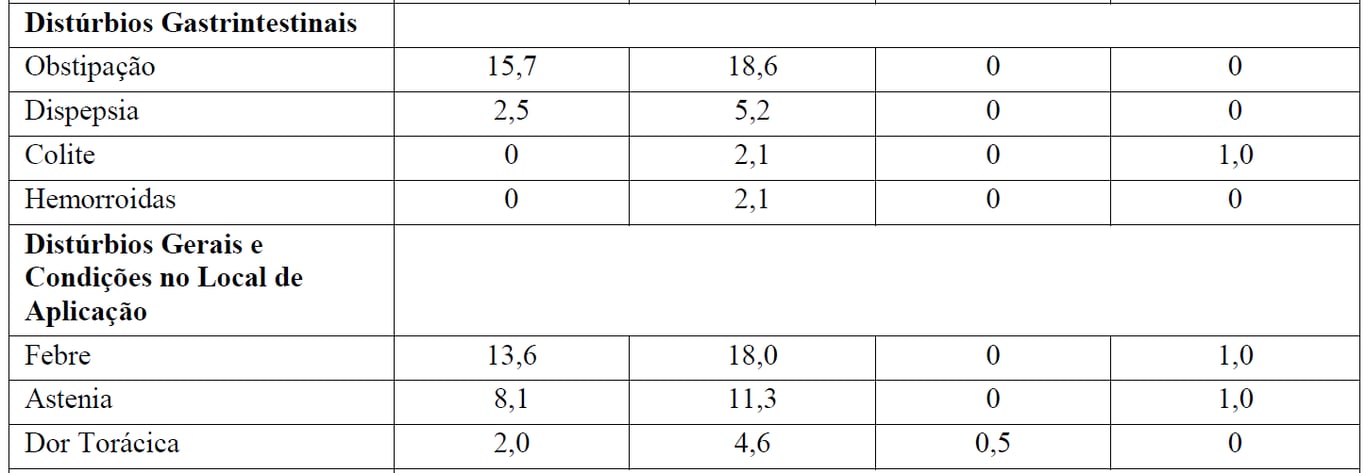
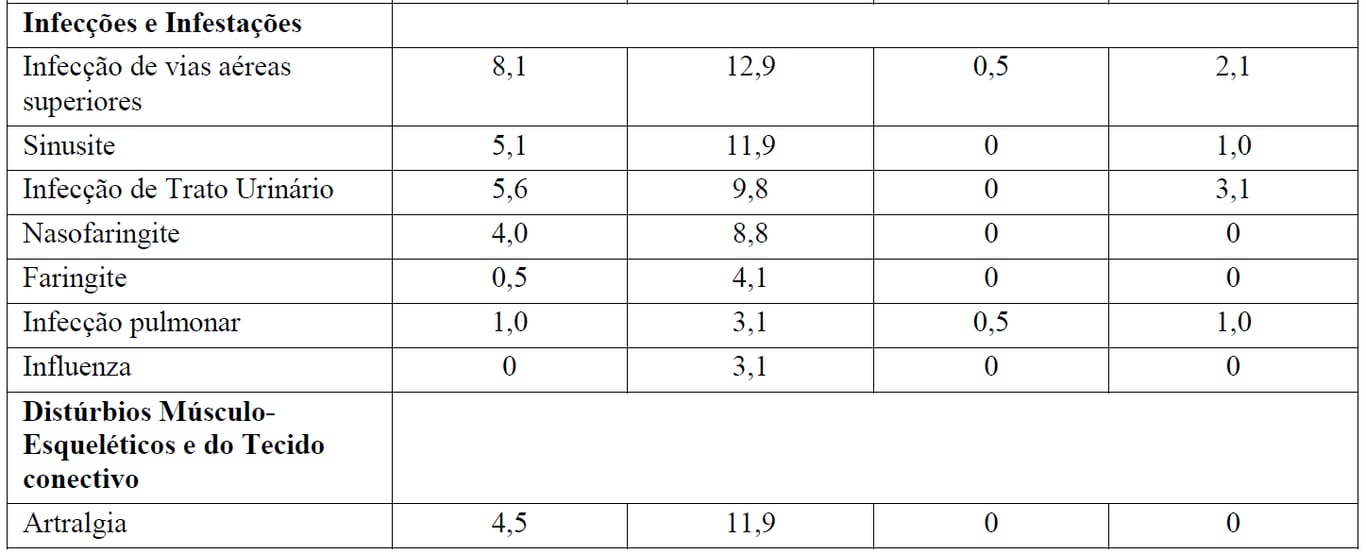
*Seguido por Gazyva manutenção.
aReações adversas codificadas pelo MedDRA conforme relato dos investigadores (excluindo as reações infusionais).
† Não foi observada nenhuma reação adversa Grau 5 com uma diferença > 2% entre os braços de tratamento.
‡ definido como qualquer evento adverso relacionado que tenha ocorrido durante ou no prazo de 24 horas da infusão.
Pacientes no braço B receberam 6 meses de tratamento de indução apenas, enquanto que depois do período de indução os pacientes no braço G+B continuaram com tratamento de manutenção com Gazyva. Durante o período de manutenção com Gazyva, as reações adversas mais comuns foram tosse (14,7%), infecções de vias aéreas superiores (11,9%), neutropenia (10,5%), sinusite (9,8%), diarreia (8,4%), reações infusionais (8,4%), náuseas (7,7%), fadiga (7,7%), bronquite (7,0%), artralgia (7,0%), nasofaringite (6,3%), infecções de trato urinário (6,3%) e febre (5,6%).
As reações adversas graus 3-5 mais comuns foram neutropenia (9,8%) e anemia, neutropenia febril, trombocitopenia, sepse, infecção de vias aéreas superiores e infecção de trato urinário (todas com 1,4%).
Informações adicionais sobre algumas reações adversas ao obinutuzumabe
Composição
Cada frasco-ampola de dose única com 40 mL contém:
Obinutuzumabe - 1000 mg (25 mg/mL).
Excipientes: Histidina, cloridrato de histidina monoidratado, trealose di-hidratada, Poloxamer 188 e água para injetáveis.
Superdosagem
Não existe nenhuma experiência com administração de dose excessiva (superdose) em estudos clínicos em seres humanos. Em estudos clínicos, foram administradas doses de Gazyva variando de 50 mg até 2000 mg por infusão. A frequência e a intensidade das reações adversas não parecem depender da dose.
Pacientes que apresentarem uma superdose devem ter sua infusão imediatamente interrompida ou reduzida e devem ser monitorados cuidadosamente. Deve-se considerar o monitoramento regular do número de linfócitos por meio de exames de sangue regulares e o risco aumentado de infecções pela redução das defesas naturais do organismo.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível. Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação Medicamentosa
Não foram realizados estudos formais de interação droga-droga. O risco de interações com outros medicamentos usados concomitantemente não pode ser descartado.
Ação da Substância
Resultados de Eficácia
Estudo clínico fase III, internacional, multicêntrico, aberto, randomizado, realizado em dois estágios, com três braços (BO21004/CLL11) para investigar o perfil de segurança e eficácia de Obinutuzumabe (substância ativa) mais clorambucil (G-Clb) em comparação com rituximabe mais clorambucil ou clorambucil (Clb) isolado foi conduzido incluindo pacientes com leucemia linfocítica crônica não tratados previamente com comorbidades.
Antes da inclusão, os pacientes precisavam apresentar LLC, CD20 positivo e uma ou ambas das seguintes medidas de condições clínicas coexistentes: escore de comorbidade (escala CIRS) maior que 6 ou função renal reduzida mensurada pela depuração de creatinina (CrCl) < 70 mL/min. Pacientes com função hepática [NCICTC Grau 3 para testes de função hepática (AST, ALT > 5 x ULN por mais de 2 semanas; bilirrubina > 3 x ULN)] e função renal (CrCl < 30 mL/min) não adequadas foram excluídos.
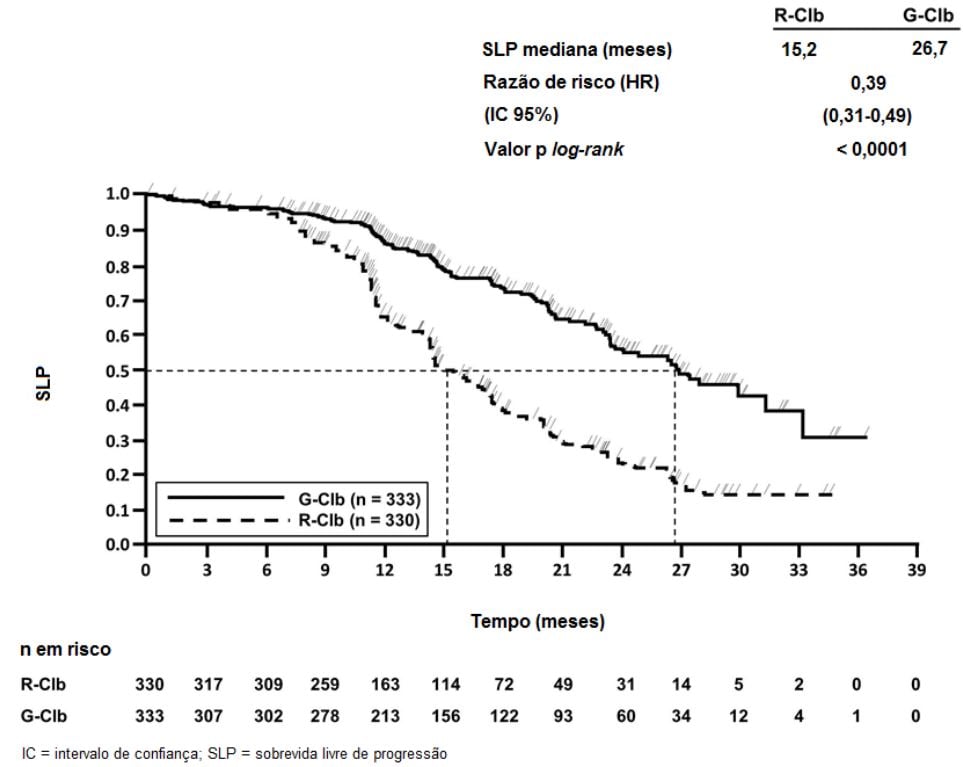
Um total de 781 pacientes foi randomizado na proporção de 2:2:1 para receber Obinutuzumabe (substância ativa) mais clorambucil, rituximabe mais clorambucil ou clorambucil isolado. O Estágio 1 comparou Obinutuzumabe (substância ativa) mais clorambucil com clorambucil isolado em 356 pacientes e o Estágio 2 comparou Obinutuzumabe (substância ativa) mais clorambucil com rituximabe mais clorambucil em 663 pacientes. Os resultados de eficácia estão resumidos na Tabela 1 e nas Figuras 1-3.
Na maioria dos pacientes, Obinutuzumabe (substância ativa) foi administrado por via intravenosa na dose inicial de 1000 mg nos Dias 1, 8 e 15 do primeiro ciclo de tratamento. Para reduzir a porcentagem de reações infusionais nos pacientes, 140 deles receberam a primeira dose de Obinutuzumabe (substância ativa) administrada em dois dias [Dia 1 (100 mg) e o Dia 2 (900 mg)]. Para cada ciclo de tratamento subsequente (Ciclos 2 a 6), os pacientes receberam 1000 mg de Obinutuzumabe (substância ativa) no Dia 1 apenas. O clorambucil foi administrado oralmente na dose de 0,5 mg/kg de peso corporal nos dias 1 e 15 de todos os ciclos de tratamento (1 a 6).
Os dados demográficos e as características basais foram bem equilibrados entre os grupos de tratamento. A maioria dos pacientes incluídos era caucasiana (95%) e do sexo masculino (61%). A mediana de idade foi de 73 anos, sendo que 44% tinham 75 anos de idade ou mais. No período basal, 22% dos pacientes apresentavam estadio A de Binet, 42% apresentavam estadio B e 36%, estadio C. O escore mediano de comorbidade foi 8 e 76% dos pacientes incluídos apresentavam escore de comorbidade acima de 6. A mediana estimada de CrCl foi de 62 mL/min e 66% de todos os pacientes apresentavam CrCl < 70 mL/min. Quarenta e dois por cento dos pacientes incluídos apresentavam tanto CrCl < 70 mL/min como um escore de comorbidade > 6. Trinta e quatro por cento dos pacientes foram incluídos no escore de comorbidade isolado e 23% com função renal comprometida somente.
As condições clínicas coexistentes mais frequentemente reportadas (usando um corte de 30% ou mais) no sistema MedDRA são: distúrbios vasculares (73%), distúrbios cardíacos (46%), distúrbios gastrintestinais (38%), distúrbios do metabolismo e nutrição (40%), distúrbios renais e urinário (38%) e distúrbios musculoesqueléticos e do tecido conectivo (33%).
O desfecho primário do estudo foi a sobrevida livre de progressão (SLP) avaliada pelo investigador (SLP-INV). Além disso, uma comissão de revisão independente (IRC) avaliou todos os pacientes com relação à progressão e avaliou a SLP (SLP-IRC).
Os principais desfechos secundários de eficácia foram o percentual de resposta no final do tratamento, a remissão molecular no final do tratamento (doença residual mínima) e o tempo até desfechos (sobrevida livre de evento, nova terapia antileucêmica). A sobrevida global para o Estágio 1 é apresentada na Figura 2. A sobrevida global para o Estágio 2 continuará sendo acompanhada e os dados são ainda imaturos.
Tabela 1. Resumo de eficácia do estudo BO21004/CLL11
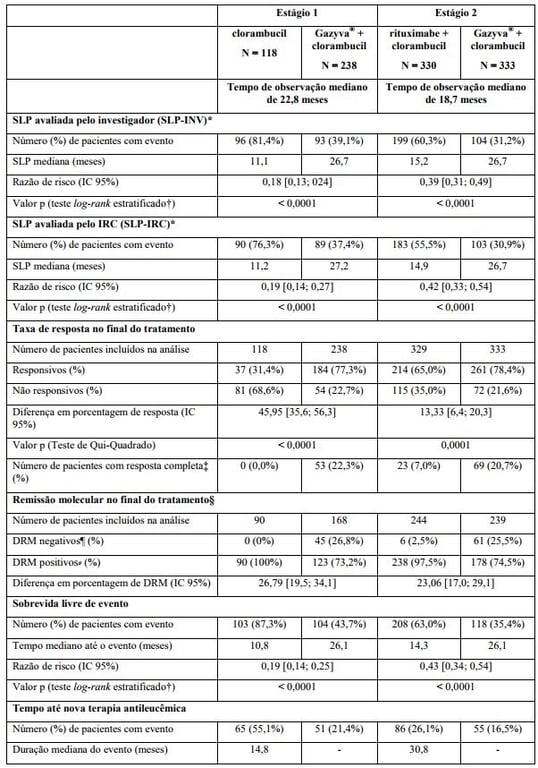
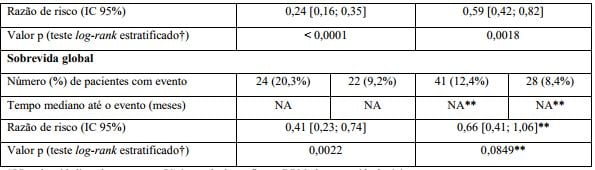
SLP: sobrevida livre de progressão; IC: intervalo de confiança; DRM: doença residual mínima.
* Definida como o tempo desde a randomização até a primeira ocorrência de progressão, recidiva ou óbito por qualquer causa conforme avaliação do investigador.
† Estratificado por estágio Binet no período basal.
‡ Inclui 11 pacientes no braço G-Clb com uma resposta completa e recuperação incompleta da medula.
§ Sangue e medula óssea combinados.
¶ Negatividade DRM definida como resultado abaixo de 0,0001.
? Inclui pacientes DRM positivos e pacientes que progrediram ou morreram antes do final do tratamento NA = não alcançado.
** Dado ainda imaturo.
Resultados da análise de SLP do subgrupo (sexo, idade, estágios de Binet, CrCl, escore CIRS, beta2-microglobulina, estado mutacional do IgVH, anormalidades cromossômicas, contagem de linfócitos no período basal) foram compatíveis com os resultados observados no total da população com intenção de tratamento (ITT). O risco de progressão da doença ou óbito (SLP) foi reduzido no braço Obinutuzumabe (substância ativa) mais clorambucil (GClb), em comparação com o braço que recebeu rituximabe mais clorambucil (RClb) e clorambucil isolado (Clb) em todos os subgrupos. A razão de risco variou de 0,08 a 0,42 para GClb vs. Clb e 0,28 a 0,71 para GClb vs. RClb.
Figura 1. Curva de Kaplan-Meier de Sobrevida Livre de Progressão do Estágio 1 Avaliada pelo Investigador
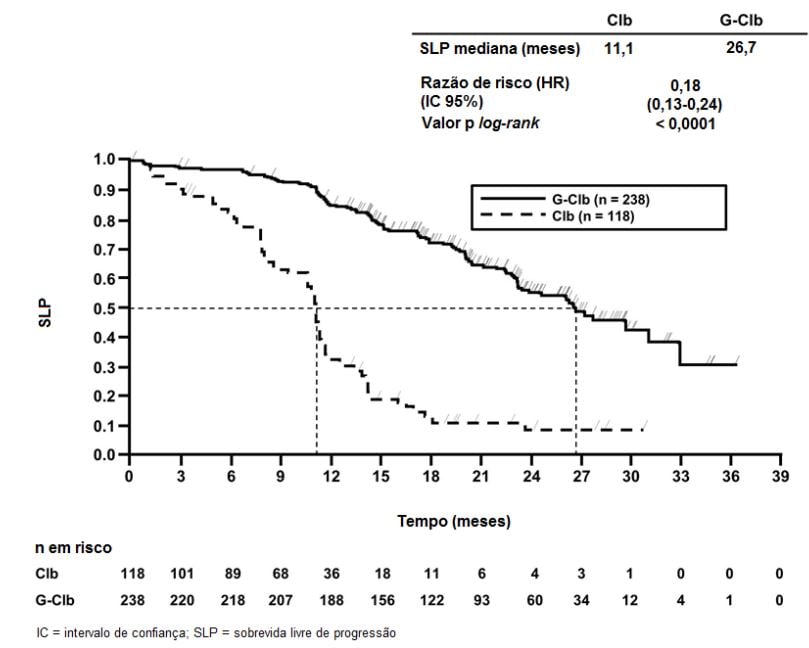
Figura 2. Curva de Kaplan-Meier de Sobrevida Global do Estágio 1
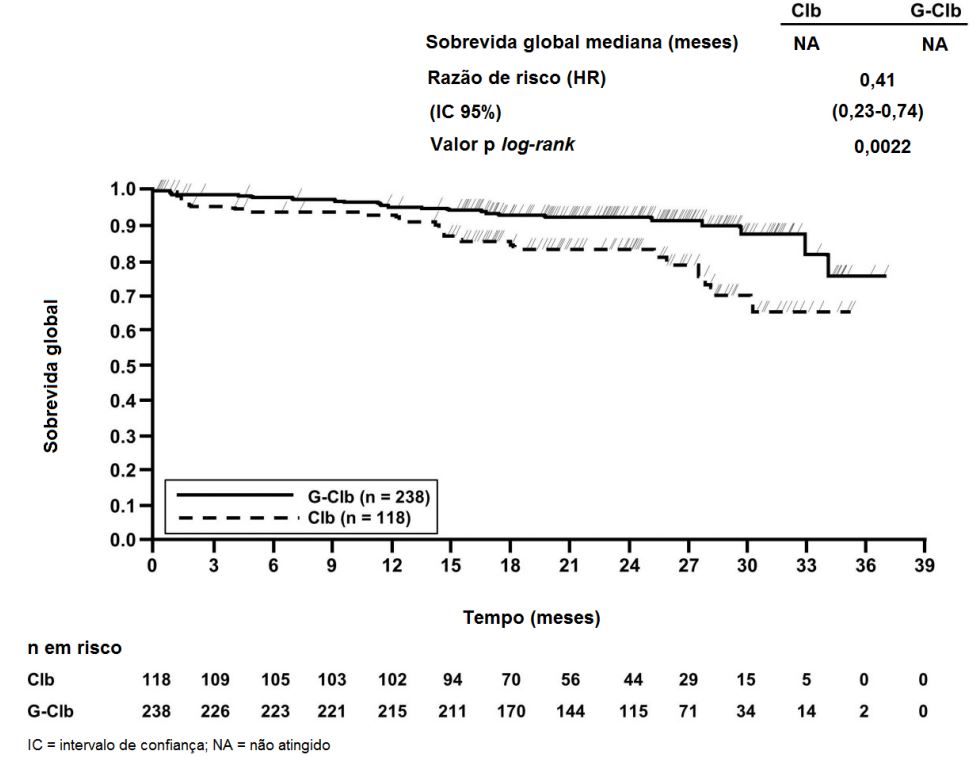
Figura 3. Curva de Kaplan-Meier de Sobrevida Livre de Progressão do Estágio 2 Avaliada pelo Investigador
Resultados Reportados pelos Pacientes
Nos questionários QLQC30 e QLQ-CLL-16, conduzidos durante o período de tratamento, não foi observada nenhuma diferença substancial em nenhuma das subescalas. Dados durante o acompanhamento, especialmente para o braço clorambucil isolado, são limitados. No entanto, nenhuma diferença notável foi identificada até o momento durante o acompanhamento do parâmetro qualidade de vida.
Avaliações de qualidade de vida relacionada à saúde, específicas para fadiga durante o período de tratamento, não mostram diferença estatisticamente significativa sugerindo que a adição de Obinutuzumabe (substância ativa) ao regime de tratamento com clorambucil não aumenta a sensação de fadiga para os pacientes.
Imunogenicidade
Pacientes no estudo pivotal BO21004/CLL11 foram testados em vários momentos para anticorpos antidrogas (ATA) contra Obinutuzumabe (substância ativa) . Nos pacientes tratados com Obinutuzumabe (substância ativa) , 8 de 140 na fase randomizada e 2 em 6 na fase de inclusão tiveram testes positivos para ATA em 12 meses de acompanhamento. Desses pacientes, nenhum apresentou reações anafiláticas ou de hipersensibilidade consideradas relacionadas aos ATA, tampouco a resposta clínica foi afetada.
Resultados de ensaio de imunogenicidade são altamente dependentes de diversos fatores, incluindo sensibilidade e especificidade do ensaio, sua metodologia, robustez a quantidade de Obinutuzumabe (substância ativa) circulante, manipulação de amostras, cronograma de coleta de amostras, medicações concomitantes e doença subjacente. Por essas razões, a comparação da incidência de anticorpos contra Obinutuzumabe (substância ativa) com a incidência de anticorpos contra outros medicamentos pode estar equivocada.
Características Farmacológicas
Propriedades Farmacodinâmicas
Propriedades Farmacocinéticas
Foi desenvolvido um modelo farmacocinético populacional para analisar dados farmacocinéticos de 678 pacientes com LNH (linfoma não Hodgkin) ou LLC que receberam Obinutuzumabe (substância ativa) nos estudos Fase I, II e III, o qual foi utilizado para descrever as características farmacocinéticas de obinutuzumabe em pacientes com LLC.
Segurança pré-clínica
Cuidados de Armazenamento
Antes de aberto, Gazyva deve ser conservado sob refrigeração (entre 2°C e 8°C).
O produto deve ser mantido na embalagem original, de forma a protegê-lo da luz. Não congelar. Não agitar.
O profissional da saúde saberá como armazenar a solução para infusão contendo Gazyva.
Número de lote e datas de fabricação e validade: Vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Característica física
Gazyva é um líquido límpido, incolor a levemente castanho, fornecido em dose única de 1000 mg em um frasco- ampola de vidro estéril, isento de conservantes, não pirogênico, contendo 40 mL de líquido concentrado (25 mg/mL).
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Todo medicamento deve ser mantido fora do alcance das crianças.
Descarte de medicamentos não utilizados e/ou com data de validade vencida
O descarte de medicamentos no meio ambiente deve ser minimizado. Os medicamentos não devem ser descartados no esgoto, e o descarte em lixo doméstico deve ser evitado. Utilize o sistema de coleta local estabelecido, se disponível.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde.
Dizeres Legais
MS-1.0100.0660
Farm. Resp.:
Tatiana Tsiomis Díaz - CRF-RJ n°. 6942
Fabricado para:
F. Hoffmann-La Roche Ltd, Basileia, Suíça
Por Roche Diagnostics GmbH, Mannheim, Alemanha
Embalado por:
F. Hoffmann-La Roche Ltd, Kaiseraugst, Suíça
Registrado, importado e distribuído no Brasil por:
Produtos Roche Químicos e Farmacêuticos S.A.
Est. dos Bandeirantes, 2020
CEP 22775-109 - Rio de Janeiro - RJ CNPJ 33.009.945/0023-39
Serviço Gratuito de Informações – 0800 7720 289
Uso retrito a hospitais.
Venda sob prescrição médica.
informações complementares
| Fabricante |
| ROCHE |
| Princípio ativo |
| Obinutuzumabe |
| Categoria do medicamento |
| Medicamentos Especiais |