para o que é indicado e para que serve?
Para que serve Asma alérgica Xolair é usado para tratamento de asma alérgica persistente moderada a grave em adultos e crianças (acima de 6 anos de idade) cujos sintomas não estão controlados por corticosteroides inalatórios (CI).Continue lendo...
ofertas de
Xolair - 150Mg 1 E 1
ofertas de Xolair - 150Mg 1 E 1
R$ 3.288,70
R$ 3.288,70
R$ 3.288,80
R$ 3.288,85
ATENÇÃO: O texto abaixo deve ser utilizado apenas como uma referência secundária. É um registro histórico da bula, rótulo ou manual do produto. Este texto não pode substituir a leitura das informações que acompanha o produto, cujo fabricante podem mudar a formulação, recomendação, modo de uso e alertas legais sem que sejamos previamente comunicados. Apenas as informações contidas na própria bula, rótulo ou manual que acompanha o produto é que devem estar atualizadas de acordo com a versão comercializada porém, no caso de qualquer dúvida, consulte o serviço de atendimento ao consumidor do produto ou nossa equipe.
Para que serve
Asma alérgica
Xolair é usado para tratamento de asma alérgica persistente moderada a grave em adultos e crianças (acima de 6 anos de idade) cujos sintomas não estão controlados por corticosteroides inalatórios (CI).
Urticária Crônica Espontânea (UCE)
Xolair (omalizumabe) é indicado como terapia adicional para uso adulto e pediátrico (acima de 12 anos de idade) em pacientes com urticária crônica espontânea refratária ao tratamento com anti-histamínicos H1.
Como o Xolair funciona?
Asma alérgica
Xolair age bloqueando uma substância chamada imunoglobulina E (também conhecida simplesmente como IgE) que é produzida pelo nosso corpo. IgE tem um papel fundamental na causa da asma alérgica.
A dosagem sanguínea de IgE deve ser medida pelo seu médico antes do início do tratamento com Xolair.
Urticária Crônica Espontânea
Xolair age bloqueando uma substância chamada imunoglobulina E (também conhecida simplesmente como IgE) que é produzida pelo nosso corpo. Como consequência, a atividade de receptores e/ou células específicas do corpo que desempenham uma função importante no aparecimento da urticária crônica espontânea é reduzida. Isto leva à redução de sintomas como coceira e lesões de urticária.
Contraindicação
Xolair é contraindicado a pacientes com alergia ao omalizumabe ou a qualquer um dos ingredientes do produto.
Se você suspeita ser alérgico, consulte seu médico.
Como usar
Xolair 150 mg é fornecido como um pó branco em um frasco-ampola com uma ampola contendo 2 mL de água para injeção. O pó deve ser dissolvido na água para injeção antes de ser injetado.
Xolair deve ser administrado por via subcutânea por uma pessoa habilitada.
Asma Alérgica
Antes de iniciar sua terapia com Xolair, seu médico deve realizar um exame de sangue para medir seu nível de IgE.
Seu médico calculará a quantidade de Xolair necessária e a frequência com que você deve tomar o medicamento. Isto depende de seu peso e da quantidade sanguínea de IgE.
Leia atentamente os itens abaixo e siga a orientação do seu médico.
Dosagem
Asma Alérgica
Você receberá 1 a 4 injeções a cada duas ou a cada quatro semanas.
Você precisará continuar utilizando seu medicamento atual para asma durante o tratamento com Xolair.
Não pare de tomar nenhum medicamento para asma sem consultar seu médico.
Urticária Crônica Espontânea
Você receberá 2 injeções de uma vez a cada quatro semanas.
Continue tomando o seu medicamento atual para UCE durante o tratamento com Xolair. Não interrompa qualquer medicamento sem o conhecimento do seu médico.
Quando usar Xolair
Asma Alérgica
Xolair será administrado a cada 2 ou 4 semanas, conforme prescrição do seu médico.
Urticária Crônica Espontânea
Você receberá Xolair uma vez a cada 4 semanas.
Por quanto tempo tomar Xolair
Continue utilizando Xolair conforme orientado pelo seu médico.
Se você tiver dúvidas por quanto tempo tomar Xolair, converse com seu médico ou farmacêutico.
Asma Alérgica
Pode ser que você não note uma melhora imediata após o início do tratamento com Xolair. Normalmente, são necessárias várias semanas para se obter o efeito desejado.
Se você parar de tomar Xolair
A interrupção ou término do tratamento com Xolair pode levar a recorrência dos sintomas de asma ou UCE.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento. Não interrompa o tratamento sem o conhecimento do seu médico.
O que devo fazer quando eu me esquecer de usar o Xolair?
Se você esquecer uma aplicação de Xolair, contate seu médico. Não use uma dose dupla para compensar a dose esquecida.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Precauções
Você não deve utilizar Xolair para tratar sintomas agudos de asma, como uma crise de asma repentina. Você deve usar um medicamento específico para este caso.
Xolair contém uma proteína e, portanto pode causar reações alérgicas graves em algumas pessoas. Uma reação alérgica grave chamada anafilaxia ocorreu em alguns pacientes após receberem Xolair. A anafilaxia pode trazer risco à vida, por isso procure imediatamente cuidados médicos caso os sintomas ocorram.
Os sinais e sintomas da anafilaxia incluem
- Chiado no peito, falta de ar, tosse, aperto no peito ou dificuldade para respirar;
- Pressão baixa, tontura, cansaço, batimento cardíaco acelerado ou fraco, ansiedade ou sensação de desconforto;
- Rubor, coceira, urticária ou sensação de calor;
- Inchaço da garganta ou língua, fechamento da garganta, rouquidão ou dificuldade de engolir.
Procure atendimento médico imediatamente se você tiver sinais e sintomas de anafilaxia após utilizar Xolair.
Anafilaxia relacionada à Xolair pode acontecer
- Logo após receber uma injeção de Xolair ou horas depois;
- Após qualquer injeção de Xolair. As anafilaxias ocorrem após a primeira injeção de Xolair ou após muitas injeções de Xolair.
Seu médico deve observá-lo quanto aos sinais e sintomas da anafilaxia após cada injeção de Xolair, por um período de tempo adequado no hospital. Se você apresentar sinais e sintomas de anafilaxia, informe seu médico imediatamente.
Seu médico deve instruí-lo a procurar tratamento médico de emergência e cuidados médicos adicionais caso você apresente os sinais e sintomas de anafilaxia após deixar o hospital.
Um tipo específico de reação alérgica (doença do soro) foi observado em pacientes tratados com Xolair ou produtos similares. Os sinais incluem dores nas articulações, rigidez, rash, febre, inchaço/aumento dos nódulos linfáticos e ocorrem geralmente dentre 1 a 5 dias após a injeção. Se você tiver uma reação como esta após usar Xolair, procure seu médico imediatamente.
Você não deve utilizar Xolair para prevenir ou tratar outras condições alérgicas do tipo
- Reações alérgicas repentinas;
- Síndrome de hiperimunoglobulina E (uma imunodeficiência herdada);
- Aspergilose (uma doença pulmonar causada por fungo);
- Alergia à comida, alergia na pele ou febre do feno.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use este medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde.
Reações Adversas
Como todos os medicamentos, pacientes tratados com Xolair podem experimentar efeitos adversos, embora nem todos os pacientes os apresentem.
Alguns efeitos adversos podem ser graves.
Efeitos adversos raros (ocorrem entre 0,01% e 0,1% dos pacientes que utilizam este medicamento)
Xolair contém uma proteína, e como qualquer proteína, potencialmente podem ocorrer reações alérgicas locais ou sistêmicas. Reações alérgicas graves repentinas têm sido raramente relatadas.
Consultar seu médico imediatamente, se você notar sinais repentinos de alergia, tais como:
- Rash (vermelhidão);
- Coceira ou urticária na pele;
- Inchaço na face, lábios, língua ou outras partes do corpo;
- Aceleração dos batimentos cardíacos;
- Tontura e sensação de cabeça vazia;
- Falta de ar;
- Chiado;
- Problemas respiratórios.
Se você tem histórico de reações alérgicas graves (anafilaxia) não relacionadas com Xolair você pode ter um risco maior de desenvolver uma reação alérgica grave após administração de Xolair.
Outros efeitos adversos que foram relatados após a introdução no mercado
- Contagem baixa de plaquetas sanguíneas com o aparecimento de sintomas como sangramento ou equimoses mais facilmente do que o normal;
- Aparecimento de alguns dos seguintes sintomas nas articulações: dor na articulação, paralisia ou formigamento nos braços e pernas, inchaço ou protuberância na pele, fraqueza e fadiga, perda de apetite e perda de peso (sinais da síndrome de Churg-Strauss);
- Aparecimento de alguns dos seguintes sintomas nas articulações: dor na articulação, rigidez, rash (vermelhidão), febre, inchaço/aumento dos nódulos linfáticos (sinais da chamada doença do soro). Quando isto ocorre, é geralmente entre o primeiro e o quinto dia após a injeção.
Se você apresentar qualquer um desses efeitos, informe seu médico imediatamente.
Alguns efeitos adversos são muito comuns – ocorrem em mais de 10% dos pacientes que utilizam este medicamento
- Dor de cabeça.
Se este efeito te afetar gravemente, avise seu médico.
Alguns efeitos adversos são comuns – ocorrem entre 1% e 10% dos pacientes que utilizam este medicamento
- Reações no local da injeção incluindo dor, inchaço, coceira e vermelhidão;
- Dor na parte superior do abdômen (em crianças);
- Febre (muito comum em crianças);
- Dor de garganta e nariz entupido (nasofaringite);
- Sensação de pressão ou dor nas bochechas e testa (sinusite e cefaleia sinusal);
- Infecção do trato respiratório superior, como inflamação da faringe e resfriado comum;
- Sensação de queimação ou dor ao urinar e necessidade frequente de urinar (possível sintoma de infecção do trato urinário);
- Dor nos membros superiores ou inferiores (braços e/ou pernas);
- Dor nos músculos e/ou ossos e/ou articulações (dor musculoesquelética, mialgia, artralgia).
Efeitos adversos incomuns – ocorrem entre 0,1% e 1% dos pacientes que utilizam este medicamento
- Sensação de tontura, sonolência ou cansaço;
- Formigamento ou entorpecimento das mãos ou pés;
- Desmaio;
- Hipotensão postural (pressão arterial baixa enquanto sentado e em pé);
- Rubor;
- Dor de garganta, tosse, problemas respiratórios agudos;
- Náusea, diarreia, indigestão;
- Coceira, urticária, rash, aumento de sensibilidade da pele ao sol;
- Aumento de peso;
- Sintomas de gripe;
- Inchaço das articulações;
- Perda de cabelo.
Se algum desses efeitos afetarem você gravemente, informe seu médico.
Se você notar quaisquer outros efeitos adversos não mencionados nesta bula, por favor, informe seu médico ou farmacêutico.
Atenção: este produto é medicamento que possui nova indicação terapêutica no país e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, informe seu médico.
População Especial
Uso pediátrico
Asma Alérgica
Não use Xolair em crianças com idade abaixo de 6 anos.
O uso de Xolair em crianças abaixo de 6 anos de idade não foi suficientemente estudado.
Urticária Crônica Espontânea
Não use Xolair em crianças menores de 12 anos.
O uso em crianças abaixo de 12 anos de idade ainda não foi suficientemente estudado.
Pacientes idosos
Xolair pode ser usado por pacientes com 65 anos ou mais.
Não há evidências que sugiram quaisquer precauções especiais necessárias para o tratamento de pacientes idosos, embora as experiências ainda sejam limitadas.
Pacientes com problemas nos rins ou fígado
Se você tem problemas nos rins ou no fígado, por favor, consulte seu médico antes de usar Xolair.
Infecções parasitárias
Se você mora em uma região onde infecções parasitárias são frequentes ou viaja para este tipo de região, por favor, avise seu médico. Xolair pode diminuir sua resistência contra essas infecções.
Se você estiver sob tratamento contra infecções parasitárias, por favor, avise seu médico. Xolair pode diminuir a eficácia de seu tratamento.
Gravidez
Se você estiver grávida ou planeja engravidar, consulte seu médico antes de iniciar o tratamento com Xolair. Seu médico irá discutir com você os benefícios e potenciais riscos de tomar este medicamento durante a gravidez.
Se você engravidar enquanto estiver utilizando Xolair, informe seu médico imediatamente.
Amamentação
Se você estiver amamentando ou pretende amamentar, consulte seu médico antes de Xolair ser administrado. Xolair pode passar do seu leite para seu bebê.
Efeitos sobre a habilidade de dirigir veículos e/ou utilizar máquinas
Xolair pode fazer você se sentir sonolento ou tonto. Se isto ocorrer, você não deve dirigir ou operar máquinas.
Composição
Cada frasco-ampola contém 150 mg de omalizumabe, um anticorpo monoclonal humanizado fabricado a partir de uma linhagem de células de mamíferos.
Excipientes: sacarose, histidina, cloridrato de histidina monoidratado e polissorbato.
Cada ampola diluente contém 2 mL de água para injeção, usada para dissolução do pó para injeção.
Xolair reconstituído contém 125 mg/mL de omalizumabe (150 mg em 1,2 mL).
Superdosagem
Se você acidentalmente tomar mais Xolair do que o prescrito, por favor, entre em contato com seu médico para maiores orientações.
Nenhum caso de superdose foi reportado.
A dose máxima tolerada de Xolair não foi determinada.
Doses únicas intravenosas de até 4.000 mg (ou 4 g) foram administradas em pacientes sem evidência de dose limite de toxicidade. A dose acumulativa mais alta administrada para pacientes foi 44.000 mg (ou 44 g) durante um período acima de 20 semanas e esta dose não resultou em qualquer efeito adverso agudo.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível. Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação Medicamentosa
Enzimas do citocromo P450, bombas de efluxo e mecanismos de ligação proteica não estão envolvidos no clearance de Omalizumabe (substância ativa), embora exista um pequeno potencial de interações droga-droga. Nenhum estudo de interação com droga formal ou vacina foi realizado com Omalizumabe (substância ativa). Não há razão farmacológica para se esperar que medicações comumente prescritas usadas no tratamento da asma ou UCE irão interagir com Omalizumabe (substância ativa).
Asma Alérgica
Em estudos clínicos, Omalizumabe (substância ativa) foi usado comumente em conjunto com corticosteroides inalatórios ou orais, beta2- agonistas inalatórios de curta e de longa ação, modificadores de leucotrienos, teofilinas e anti-histamínicos orais. Não houve indicação que a segurança de Omalizumabe (substância ativa) foi alterada com estas outras medicações comumente usadas para asma. Dados da utilização de Omalizumabe (substância ativa) em combinação com terapia de hipossensibilização em asma sazonal são limitados.
Estão disponíveis dados de eficácia e segurança de um estudo multicêntrico DB PC da Alemanha com Omalizumabe (substância ativa) em combinação com imunoterapia específica (Depigoid) comparado à imunoterapia (IT) isolada em 132 adultos e adolescentes com asma alérgica sazonal e rinoconjuntivite alérgica sazonal associada. A população do estudo foi definida como pacientes com o diagnóstico de asma alérgica sazonal a pólen de grama (e/ou pólen de centeio) não adequadamente controlada com concomitante rinoconjuntivite alérgica sazonal em > 2 estações prévias. A duração do tratamento do estudo foi de 18 semanas no total (10 semanas de pré-estação e 8 semanas durante a estação com pólen de grama).
O objetivo primário foi a redução na carga do sintoma (soma da média diária da pontuação de gravidade do sintoma adicionado à média diária do escore de escalonamento de medicação ou uso de medicação de resgate) combinado para asma e rinoconjuntivite.
Os objetivos secundários incluíram a avaliação do investigador e do paciente da efetividade global do tratamento (GETE), Qualidade de Vida (QoL) relacionada à asma/rinite, função pulmonar, sintomas de asma e eventos adversos.
A combinação de terapia reduziu a carga de sintomas na estação com pólen de grama em 39% (p < 0,05%) em relação ao tratamento com IT isolada. Esta diferença foi devido à melhora na gravidade dos sintomas alérgicos (p = 0,01), enquanto nenhuma diferença entre os dois grupos de tratamento foi observada pela análise do escore de medicação devido ao reduzido uso de medicação de resgate nos dois grupos. A maioria dos objetivos secundários mostrou resultados significantemente melhores quando comparados a IT isolada (particularmente GETE, escores de QoL em rinite ou asma).
Urticária Crônica Espontânea
Em estudos clínicos sobre UCE, Omalizumabe (substância ativa) foi usado em combinação com anti-histamínicos (anti-H1, anti-H2) e antagonistas do receptor de leucotrienos (LTRAs). Nos estudos de Fase III Q4881g e Q4882g, todos os pacientes receberam anti-histamínicos H1, além de Omalizumabe (substância ativa) ou placebo. No estudo de Fase III Q4883g, todos os pacientes receberam um ou mais anti-histamínico(s) H1 e/ou anti-histamínicos H2 e/ou LTRAs, além de Omalizumabe (substância ativa) ou placebo. Não foi observada nenhuma evidência de que a segurança de Omalizumabe (substância ativa) tenha sido alterada quando ele foi utilizado com estes medicamentos em relação ao seu perfil de segurança conhecido em asma alérgica. Além disso, uma análise farmacocinética da população não revelou nenhum efeito relevante de anti-histamínicos H2 e LTRAs na farmacocinética do Omalizumabe (substância ativa).
O uso de Omalizumabe (substância ativa) em combinação com terapias imunossupressoras ainda não foi estudado.
Incompatibilidades
Omalizumabe (substância ativa) não deve ser misturado a qualquer outro medicamento ou diluente diferente da água para injeção.
Ação da Substância
Asma Alérgica
Resumo dos estudos A2208 e A2210
A extensão da tabela posológica para as novas combinações de IgE e peso corporal é suportada por estudos clínicos.
O estudo A2210 foi um estudo de 16 semanas, multicêntrico, randomizado, duplo-cego, de grupos paralelos e placebo controlado, em 50 pacientes adultos (18-65 anos) com asma intermitente, asma persistente leve ou asma persistente moderada. Os pacientes foram agrupados por níveis de IgE baixos (30-300 UI/mL), altos (700-2.000 UI/mL) e randomizados (2:1) para Omalizumabe (substância ativa) subcutâneo até 600 mg a cada 2 semanas (grupos de IgE baixo e alto) ou 4 semanas (grupo de IgE baixo apenas) ou placebo.
O estudo A2208 foi um estudo de segurança multicêntrico, aberto, de grupo paralelo em 32 pacientes com asma leve a moderada que receberam duas injeções subcutâneas únicas de 450 mg, 525 mg ou 600 mg de Omalizumabe (substância ativa) com 14 dias de intervalo.
Principais resultados de eficácia
No estudo A2210, Omalizumabe (substância ativa) reduziu a resposta asmática primária induzida por alérgenos (RAP, desfecho primário) em ambos os grupos baixo IgE e alto IgE comparados com placebo na semana 16, a diferença na redução de % máxima em VEF1 entre Omalizumabe (substância ativa) e placebo foi de -14,9% (p < 0,001) comparado com -8,2% (p = 0,087) no grupo de IgE baixo. [10] No estudo A2208, a diminuição máxima média de cada paciente de IgE livre na triagem foi de ? 99,0% para todas as três dosagens e a média das concentrações de IgE livre permaneceu abaixo de 25 ng/mL por pelo menos 2 semanas após a segunda dose. Reduções na IgE livre foram consistentes com os níveis previamente demonstrados a estarem associados à eficácia clínica.
Principais resultados de segurança
No estudo A2210, a frequência de reações adversas foi similar em ambos os grupos de tratamento, a maioria dos quais não são suspeitos de estarem relacionados com o medicamento.
No estudo de segurança A2208, 26 (81,3%) pacientes reportaram um total de 69 reações adversas. Destas, 10 reações adversas reportadas por 6 (18,8%) pacientes dentre todos os grupos de doses foram consideradas estarem relacionadas ao Omalizumabe (substância ativa). Estas reações adversas foram principalmente de intensidade leve, mais frequentemente causaram distúrbios no sistema nervoso ou distúrbios gastrointestinais e foram resolvidos até o final do estudo. Nenhuma das medidas laboratoriais de segurança, sinais vitais, gravações do ECG ou resultados espirométricos revelaram que quaisquer achados clínicos relevantes ou tendências podem estar relacionados com Omalizumabe (substância ativa).
No geral, os resultados dos estudos de extensão da dosagem da tabela posológica são consistentes com os perfis de segurança e eficácia já conhecidos de Omalizumabe (substância ativa).
Urticária Crônica Espontânea (UCE)
O programa de desenvolvimento clínico de Fase III para UCE incluiu três estudos randomizados, duplo-cegos, controlados por placebo, de grupos paralelos e multicêntricos: Q4881g, Q4882g e Q4883g.
Os estudos Q4881g e Q4882g avaliaram a eficácia e a segurança da administração de Omalizumabe (substância ativa) 75 mg, 150 mg ou 300 mg a cada quatro semanas durante 24 e 12 semanas, respectivamente, com um período de acompanhamento de 16 semanas sem tratamento, em pacientes (12 a 75 anos) com UCE refratária apesar do tratamento com anti-histamínico H1.
O estudo Q4883g avaliou a segurança e a eficácia de Omalizumabe (substância ativa) 300 mg administrado a cada quatro semanas durante 24 semanas, com um período de acompanhamento de 16 semanas sem tratamento em pacientes (12 a 75 anos) com UCE refratária apesar do tratamento com anti-histamínico H1 e/ou H2 e/ou antagonista do receptor de leucotrienos (LTRA).
Tabela - 1 Desfechos de Eficácia:
| Alteração da baseline até a semana 12 no Escore de Intensidade da Coceira semanal (ISS, faixa 0-21) | Desfecho primário nos estudos Q4881g e Q4882g Desfecho secundário no estudo de segurança Q4883g |
| Tempo até a resposta MID (diminuição?5 pontos em relação à baseline) no escore da intensidade da coceira semanal (ISS) até a semana 12 |
Desfechos secundários nos três estudos Q4881g, Q4882g e Q4883g |
| Alteração da baseline até a semana 12 no Escore de Atividade da Urticária durante um período de 7 dias (UAS7 b, faixa 0-42) | |
| Proporção de pacientes com Escore de Atividade da Urticária durante um período de 7 dias?6 (UAS7 b?6) na semana 12 | |
| Proporção de pacientes com Escore de Atividade da Urticária durante um período de 7 dias = 0 (UAS7 b = 0) na semana 12 c | |
| Alterações em relação à baseline no escore de número de lesões de urticária semanal na semana 12 | |
| Alteração da baseline até a semana 12 no Índice de Qualidade de Vida em Dermatologia (DLQI) geral | |
| Proporção de pacientes com dias sem angioedema da semana 4 até a semana 12 |
a MID: Diferença Minimamente Importante.
b UAS7: Composto de intensidade da coceira e número de lesões de urticária medidas diariamente e totalizados ao longo de uma semana.
c Análise post hoc do estudo Q4882g.
d A proporção média de dias sem angioedema da semana 4 até a semana 12 foi calculada para toda a população estudada, incluindo os pacientes assintomáticos quanto ao angioedema.
Nos estudos Q4881g e Q4882g, a dose de 75 mg não atendeu de forma consistente ao desfecho primário de eficácia (alteração da baseline até a semana 12 no escore de intensidade da coceira semanal) ou a vários desfechos secundários. Essa dose foi considerada sem eficácia e, consequentemente, não será mais apresentada.
Alteração do basal até a semana 12 no escore de intensidade da coceira semanal
O desfecho primário de eficácia, alteração do basal até a semana 12 no escore de intensidade da coceira semanal, foi atendido pelas doses de 150 mg e 300 mg nos estudos Q4881g e Q4882g, e pela dose de 300 mg no Q4883g (desfecho secundário, consulte a Tabela 2).
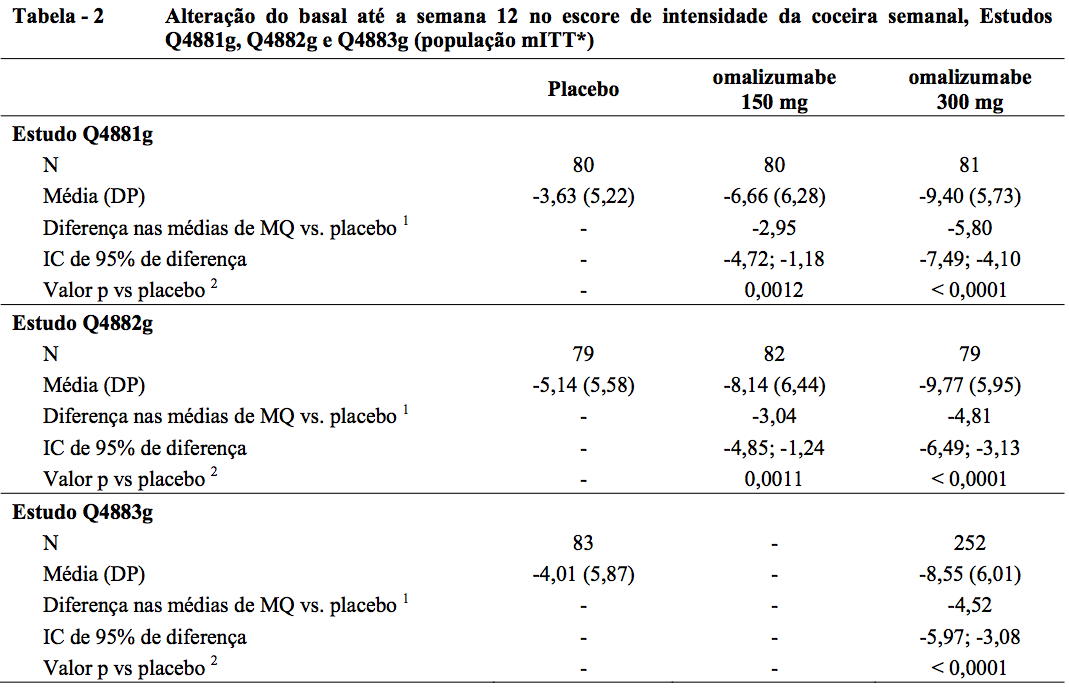
* População de Intenção de Tratamento modificada (mITT): Incluiu todos os pacientes que foram randomizados e receberam pelo menos uma dose da medicação em estudo.
BOCF (Observação de Baseline Realizada) foi usada para imputar dados ausentes.
1 A média de MQ foi estimada por meio de um modelo ANCOVA. Os estratos foram pontuação de intensidade da coceira semanal na baseline (< 13 vs. ? 13) e peso na baseline (< 80 kg vs. ? 80 kg).
2 O valor p é derivado do teste-t ANCOVA.
A Figura 1 mostra o escore médio de intensidade da coceira semanal no decorrer do tempo no estudo Q4881g. Os escores médios de intensidade da coceira semanais diminuíram de maneira significativa nos dois grupos de tratamento, com um efeito máximo em torno da semana 12 que foi sustentado durante o período de tratamento de 24 semanas. Nos estudos Q4883g (300 mg durante o período de tratamento de 24 semanas) e Q4882g (150 mg e 300 mg durante o período de tratamento de 12 semanas), os resultados foram semelhantes aos do estudo Q4881g.
Nos três estudos (consulte a Figura 1 para o estudo Q4881g), o escore médio de intensidade da coceira semanal para as duas doses aumentou gradualmente durante o período de acompanhamento de 16 semanas sem tratamento, o que é compatível com a recorrência dos sintomas. Os valores médios no final do período de acompanhamento foram semelhantes aos do grupo de placebo, mas menores que os respectivos valores médios de baseline.
BOCF = observação de baseline realizada; mITT = população de intenção de tratamento modificada.
Tempo até a diferença significativa mínima de resposta (MID) de 5 pontos no ISS semanal até a semana 12
Nos estudos Q4881g e Q4882g, os tempos para se obter uma MID de 5 pontos no escore de intensidade da coceira semanal foram estatisticamente menores de forma significativa para os pacientes no grupo de tratamento com 300 mg, em comparação com grupos de placebo com valor p < 0,0001. Um tempo menor também foi observado para os grupos de tratamento com 150 mg em comparação com placebo com p = 0,0301 no estudo Q4881g e p = 0,0101 no estudo Q4882g. Os tempos médios para alcançar a resposta MID foram de 1 semana no grupo de tratamento com 300 mg, 2 semanas nos grupos com 150 mg e 4 semanas para o placebo. Resultados semelhantes foram observados no estudo Q4883g, com tempo médio até a resposta MID de 2 semanas no grupo de tratamento com 300 mg (p < 0,0001) vs. 5 semanas no grupo de placebo.
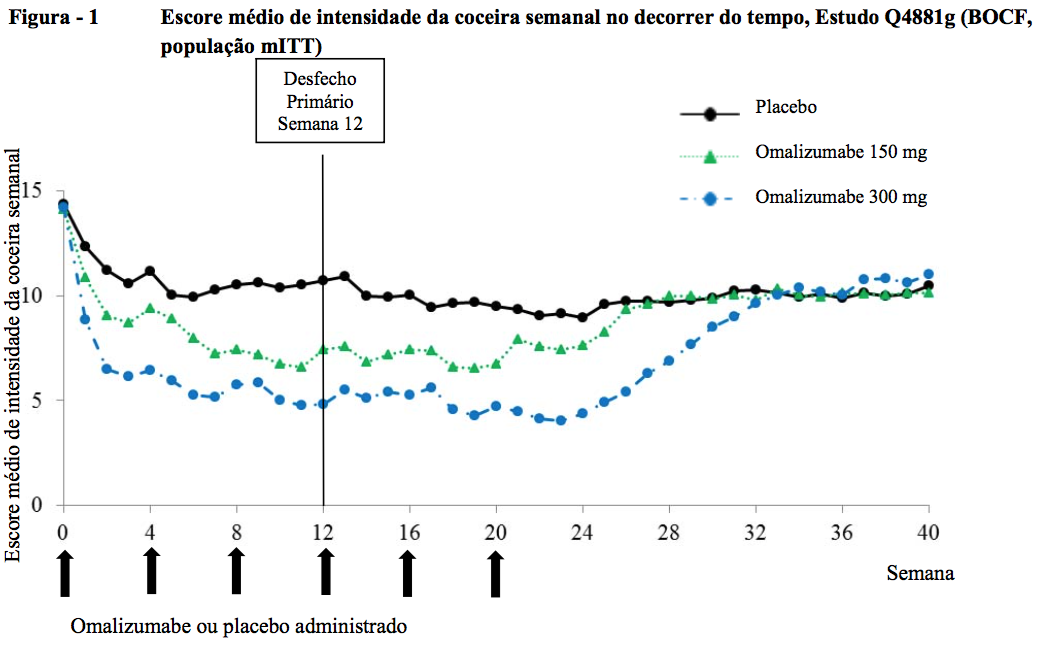
Alteração da baseline até a semana 12 no UAS7
Nos estudos de Fase III, os grupos de tratamento com Omalizumabe (substância ativa) 150 mg e 300 mg mostraram uma diferença estatisticamente significativa em relação ao placebo na alteração média da baseline até a semana 12 no UAS7 (Figura 2 para o estudo Q4881g). A significância estatística (p < 0,0001) foi alcançada nos três estudos para o grupo de tratamento com 300 mg, e nos estudos Q4881g (p = 0,0008) e Q4882g (p = 0,0001) para o grupo de tratamento com 150 mg.
A Figura 2 mostra o UAS7 médio no decorrer do tempo no estudo Q4881g, exibindo uma diminuição significativa em relação à baseline nos dois grupos de tratamento com um efeito máximo em torno da semana 12. A magnitude do efeito foi mantida durante o período de tratamento de 24 semanas. Nos estudos Q4882g (150 mg e 300 mg durante o período de tratamento de 12 semanas) e Q4883g (300 mg durante o período de tratamento de 24 semanas), os resultados foram semelhantes aos do estudo Q4881g.
Nos três estudos (consulte a Figura 2 para o estudo Q4881g), o UAS7 para os dois grupos de tratamento com Omalizumabe (substância ativa) aumentou gradualmente durante o período de acompanhamento de 16 semanas sem tratamento, o que é compatível com a recorrência dos sintomas. Os valores médios no final do período de acompanhamento foram semelhantes aos do grupo de placebo, mas menores que os respectivos valores médios de baseline.
BOCF = observação de baseline realizada;
mITT = população de intenção de tratamento modificada;
UAS7 = escore de atividade da urticária durante 7 dias.
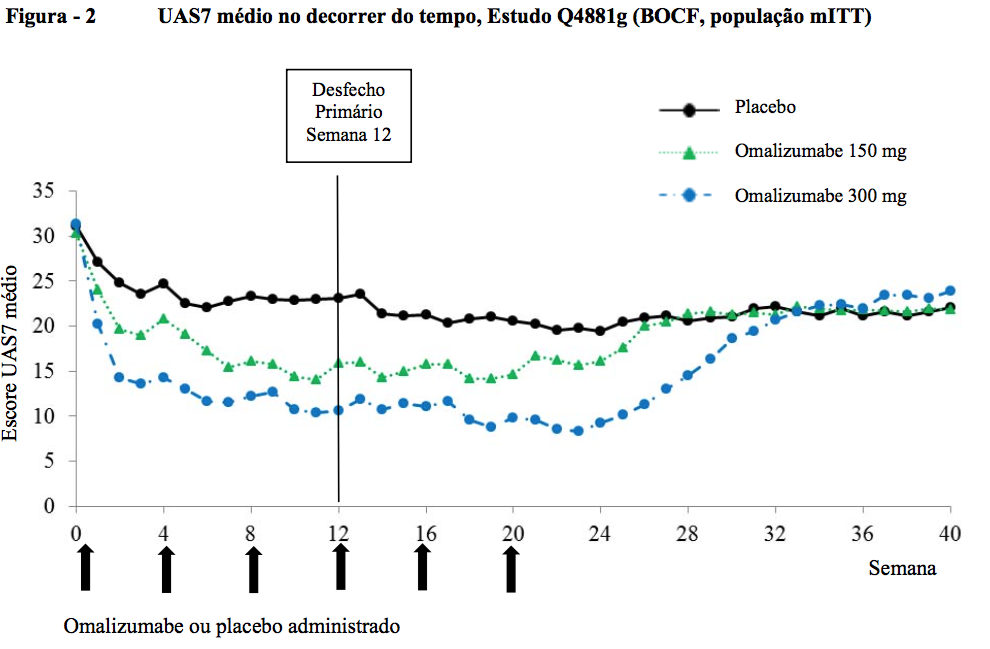
Proporção de pacientes com UAS7 ? 6 na semana 12
As taxas de resposta de UAS7 ? 6 na semana 12, variando de 52 a 66% para o grupo de tratamento com 300 mg (51,9% no Q4881g, 65,8% no Q4882g e 52,4% no Q4883g) foram todas estatisticamente maiores de forma significativa em comparação com 11 a 19% para o grupo de placebo (11,3% no Q4881g, 19,0% no Q4882g e 12,0% no Q4883g; todas com p < 0,0001). Nos grupos de tratamento com 150 mg, a proporção de pacientes com UAS7 ? 6 na semana 12, variando de 40-43% (40,0% em Q4881g, 42,7% em Q4882g) demonstrou uma diferença clinicamente notável em relação aos grupos tratados com placebo (11,3% e 19,0%; p < 0,0001 e p = 0,0010, respectivamente).
As proporções de pacientes com UAS7 ? 6 na semana 12 são apresentadas na Figura 3. As taxas de resposta variaram de 52 a 66% (dose de 300 mg) todas foram estatisticamente maiores de forma significativa em comparação com 11-19% no grupo de placebo (p < 0,0001).
As taxas de resposta para dose de 150 mg mostra uma diferença notável (40-43%; p ? 0.001) em comparação com o placebo.
Os valores p são do grupo de Omalizumabe (substância ativa) vs placebo.
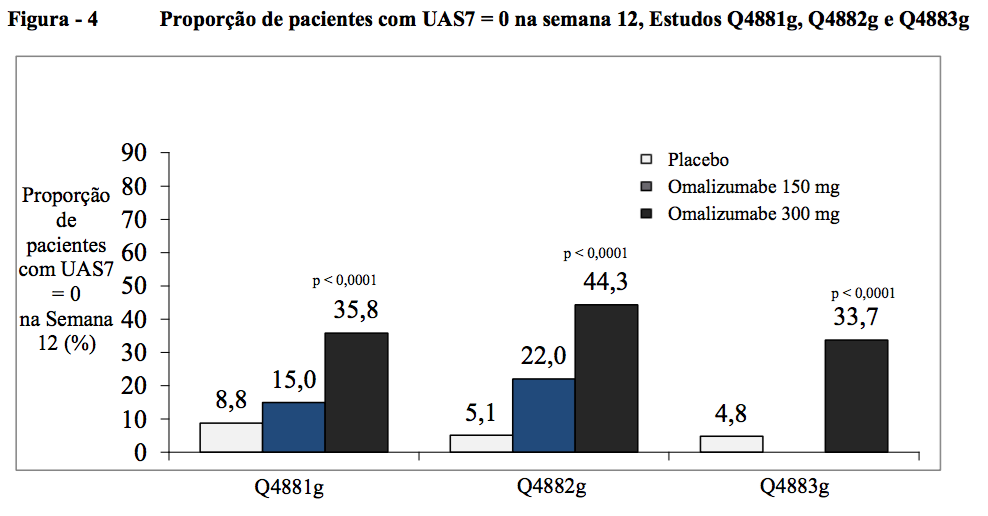
Proporção de pacientes com UAS7 = 0 na semana 12
A proporção de pacientes com resposta completa, definida por um UAS7 = 0 na semana 12, foi estatisticamente significativa para os grupos de tratamento com 300 mg em comparação ao placebo, variando de 34 a 44% (35,8% no Q4881g, 44,3% no Q4882g e 33,7% no Q4883g, contra 8,8% no Q4881g, 5,1% no Q4882g e 4,8% no Q4883g com placebo; todos p < 0,0001). Isso foi numericamente melhor para o grupo de tratamento com 150 mg, com 15,0% no Q4881g e 22,0% no Q4882g em comparação com placebo.
A proporção de pacientes com resposta completa demonstrada por um UAS7 = 0 na semana 12 variou de 34 a 44% (dose de 300 mg, estatisticamente significativa, todos p < 0,0001) em comparação com 5 a 9% no grupo de placebo. Nos grupos de tratamento com 150 mg foi observada uma diferença clinicamente notável em comparação com o placebo, variando de 15-22% (Figura 4).
Alterações em relação à baseline no escore de número de lesões de urticária semanal na semana 12
Nos três estudos de Fase III, a diferença do placebo nas alterações médias em relação à baseline no escore de número de lesões de urticária semanal na semana 12 para os grupos de tratamento com 300 mg foi estatisticamente significativa, exibindo uma diminuição no escore de número de lesões de urticária em comparação com o placebo (-11,35 no Q4881g, -11,97 no Q4882g e -10,46 no Q4883g versus -4,37; -5,22 e -4,49 para os grupos de placebo correspondentes; todos p < 0,0001). Para os grupos de tratamento com 150 mg, as alterações médias foram -7,78 (p = 0,0017) no Q4881g e -9,75 (p < 0,001) no Q4882g.
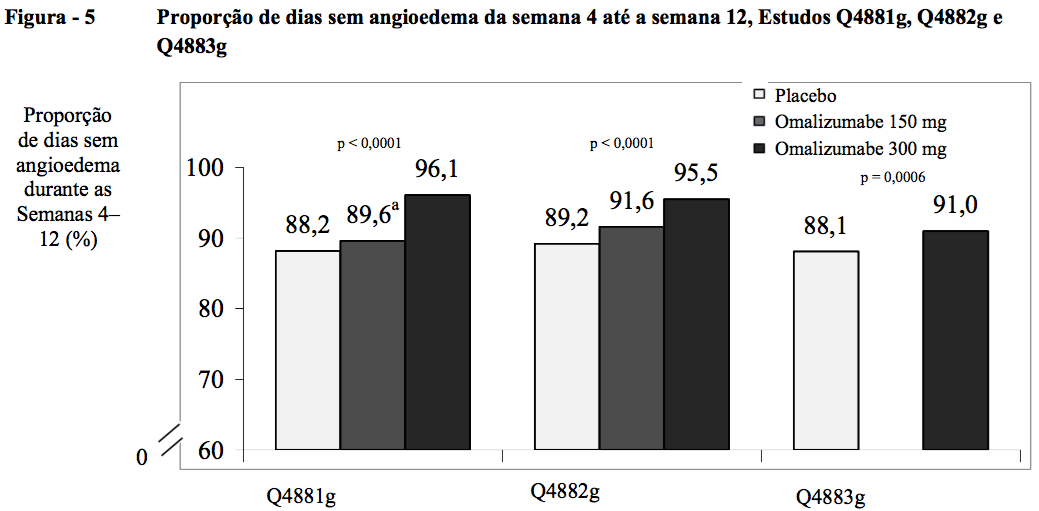
Proporção de dias sem angioedema da semana 4 até a semana 12
Nos três estudos de Fase III, os grupos de tratamento com 300 mg consistentemente obtiveram a maior proporção média de dias sem angioedema da semana 4 até a semana 12 (96,1% no Q4881g; 95,5% no Q4882g; 91% no Q4883g) em comparação com o grupo de placebo (88,2%; 89,2%; 88,1%, respectivamente; todos p < 0,0001). Nos grupos de tratamento com 150 mg, as proporções médias de dias sem angioedema no mesmo período de tempo para os estudos Q4881g e Q4882g foram 89,6% e 91,6% respectivamente, sem diferença estatisticamente significativa em relação ao placebo.
Nos três estudos de Fase III, os grupos de tratamento com 300 mg consistentemente obtiveram a maior proporção média de dias sem angioedema da semana 4 até a semana 12 (91 a 96%). O aumento na proporção de dias sem angioedema em comparação com o grupo de placebo foi estatisticamente significativo (p < 0,001) (Figura 5). No grupo de tratamento com 150 mg, as proporções médias de dias sem angioedema no mesmo período de tempo para os estudos Q4881g e Q4882g foram 89,6% e 91,6%, respectivamente. Os valores correspondentes para o placebo nos mesmos estudos foram 88,2% e 89,2%. Nestes dois estudos, as diferenças em relação ao placebo não tiveram significância estatística para a dose de 150 mg.
Os valores p são do grupo de Omalizumabe (substância ativa) vs placebo.
a Não avaliado quanto à significância estatística de acordo com o plano de controle de erro tipo I.
A proporção média de dias sem angioedema da semana 4 até a semana 12 foi calculada para toda a população estudada, incluindo os pacientes assintomáticos quanto ao angioedema.
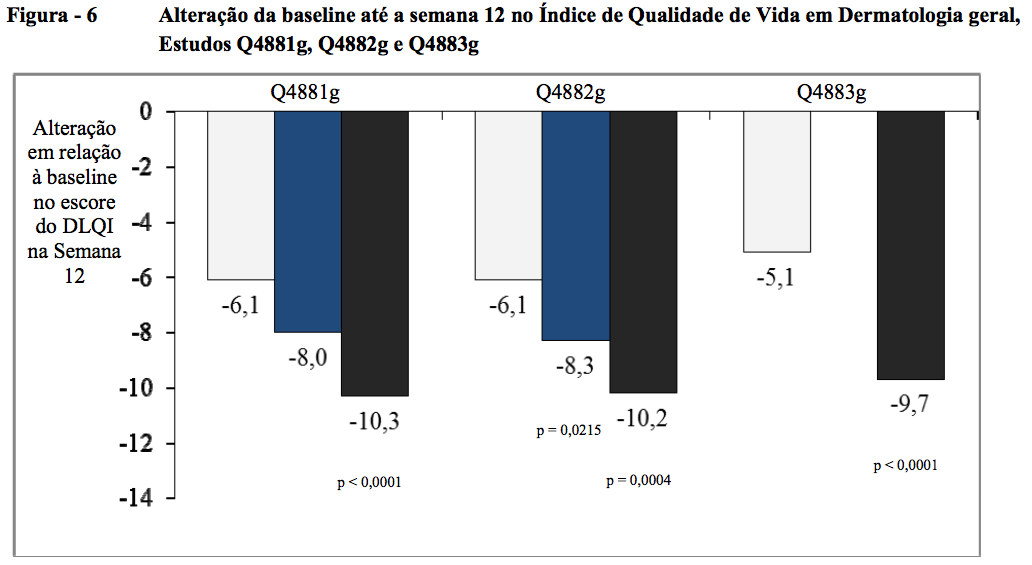
Alteração da baseline até a semana 12 no Índice de Qualidade de Vida em Dermatologia (DLQI) geral
Nos três estudos de Fase III, a alteração média da baseline até a semana 12 no DLQI geral foi significativamente maior do ponto de vista estatístico para os grupos de tratamento com 300 mg do que para os grupos com placebo, exibindo uma melhora de 10,3 pontos no Q4881g, 10,2 no Q4882g e 9,7 no Q4883g versus 6,1; 6,1 e 5,1 para os grupos de placebo correspondentes (todos p < 0,001). Para os grupos de tratamento com 150 mg, as alterações médias foram de 8,0 pontos (p = 0,2286) no Q4881g e de 8,3 pontos (p = 0,0215) no Q4882g versus 6,1 para cada um dos grupos de placebo correspondentes.
Nos três estudos de Fase III, a alteração da baseline até a semana 12 no DLQI geral foi significativamente maior (p < 0,001) do ponto de vista estatístico para o grupo de tratamento com 300 mg em comparação com o placebo. O grupo de Omalizumabe (substância ativa) 150 mg mostrou uma diferença clinicamente notável em relação ao placebo no estudo Q4882g (p = 0,022) (Figura 6 [12]).
DLQI = Índice de Qualidade de Vida em Dermatologia.
Os valores p são do grupo de Omalizumabe (substância ativa) vs placebo.
Eficácia depois de 24 semanas de tratamento
A Tabela 3 mostra os resultados depois de 24 semanas de tratamento. Magnitudes de resposta semelhantes são observadas da mesma forma em 12 semanas.
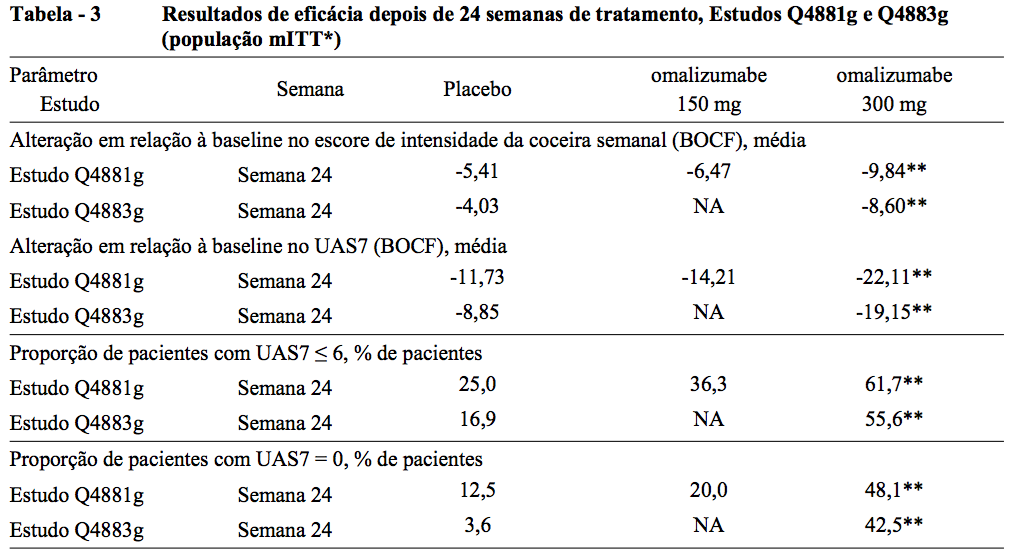
** Valor p ? 0,0001 da estatística de teste correspondente entre o tratamento e o placebo NA: Não aplicável.
BOCF: Observação de Baseline Realizada.
Características Farmacológicas
Grupo farmacoterapêutico: outras drogas sistêmicas para doenças obstrutivas das vias respiratórias, código ATC: R03DX05.
Farmacodinâmica
O Omalizumabe (substância ativa) é um anticorpo monoclonal humanizado derivado de DNA recombinante que se liga seletivamente à imunoglobulina E (IgE). O anticorpo é uma IgG1 kappa que contém regiões de estrutura humana com regiões determinantes complementares de um anticorpo murino humanizado que se liga à IgE.
Pacientes com Asma Alérgica
A cascata alérgica inicia-se quando a IgE ligada aos receptores Fc?RI de alta afinidade na superfície dos mastócitos e basófilos, sofre ligação cruzada por um alérgeno. Isto resulta na degranulação destas células efetoras e na liberação de histaminas, leucotrienos, citocinas e outros mediadores. Estes mediadores são responsáveis pela fisiopatologia da asma alérgica, incluindo edema das vias respiratórias, contração do músculo liso e alteração da atividade celular associada ao processo inflamatório. Eles também contribuem para os sinais e sintomas da doença alérgica, tais como broncoconstrição, produção de muco, sibilos, dispneia, opressão torácica, congestão nasal, espirros, prurido, rinorreia e prurido nasal e lacrimejamento.
O Omalizumabe (substância ativa) liga-se à IgE e evita a sua ligação ao receptor Fc?RI, reduzindo assim a quantidade de IgE livre que está disponível para desencadear a cascata alérgica. O tratamento de indivíduos atópicos com Omalizumabe (substância ativa) resultou em uma marcante diminuição do número de receptores Fc?RI em basófilos. Além disso, a liberação de histamina in vitro dos basófilos isolados de indivíduos tratados com Omalizumabe (substância ativa) foi reduzida em aproximadamente 90% após estimulação com um alérgeno, comparado aos valores de pré-tratamento.
Em estudos clínicos, níveis séricos de IgE livre foram reduzidos de forma dose-dependente em uma hora após a primeira dose e mantidos entre as doses. A redução média da IgE sérica livre foi maior do que 96% usando doses recomendadas. Níveis séricos de IgE total (ou seja, ligada e não ligada) aumentaram após a primeira dose devido à formação do complexo Omalizumabe (substância ativa):IgE que tem uma taxa de eliminação mais lenta comparada com a IgE livre. Nas 16 semanas após a primeira dose, a média dos níveis séricos de IgE total foi 5 vezes mais alta em relação aos níveis de pré-tratamento quando usados ensaios padrões. Após a interrupção da administração de Omalizumabe (substância ativa), o aumento de IgE total e a redução de IgE livre induzidos por Omalizumabe (substância ativa) foram reversíveis, sem rebote observado nos níveis de IgE após remoção da droga. Níveis de IgE total não retornaram aos níveis de pré-tratamento por até um ano após a descontinuação de Omalizumabe (substância ativa).
Pacientes com Urticária Crônica Espontânea (UCE)
Existem várias teorias quanto à etiologia da UCE, inclusive uma que sugere uma origem autoimune. Autoanticorpos anti-IgE e seu receptor, Fc?RI, foram isolados a partir do soro de alguns pacientes com UCE. Estes autoanticorpos podem ativar basófilos ou mastócitos, levando à liberação de histamina.
Uma hipótese sobre o mecanismo de ação do Omalizumabe (substância ativa) na UCE é que ele diminui os níveis de IgE livre no sangue e subsequentemente na pele. Isto ocasiona a sub-regulação dos receptores de IgE de superfície, diminuindo assim a sinalização a jusante através da via Fc?RI, o que resulta na supressão da ativação celular e em respostas inflamatórias. Como consequência, ocorre a redução da frequência e da intensidade dos sintomas de UCE. Outra hipótese é a de que a diminuição dos níveis de IgE livre circulante leva a uma dessensibilização rápida e não específica de mastócitos cutâneos. A sub-regulação de Fc?RI pode ajudar a sustentar a resposta.
Em estudos clínicos em pacientes com UCE, o tratamento com Omalizumabe (substância ativa) levou a uma redução dose-dependente na IgE livre e a um aumento nos níveis de IgE total no soro, de modo semelhante às observações em pacientes com asma alérgica. A supressão máxima de IgE livre foi observada 3 dias após a primeira dose subcutânea. Após a administração repetida uma vez a cada quatro semanas, os níveis séricos de IgE livre pré-dose permaneceram estáveis entre 12 e 24 semanas de tratamento. Os níveis de IgE total no soro aumentaram após a primeira dose devido à formação de complexos de Omalizumabe (substância ativa): IgE que apresentam uma taxa de eliminação mais lenta em comparação com a IgE livre. Após a administração repetida uma vez a cada quatro semanas com doses de 75 mg a 300 mg, os níveis séricos médios de IgE total pré-dose na semana 12 foram de duas a três vezes mais altos em comparação com os níveis pré-tratamento, e permaneceram estáveis entre 12 e 24 semanas de tratamento. Após a descontinuação de Omalizumabe (substância ativa), os níveis de IgE livre aumentaram e os níveis de IgE total diminuíram até os níveis pré-tratamento durante um período de acompanhamento de 16 semanas sem tratamento.
Farmacocinética
Pacientes com Asma Alérgica
Características em populações de pacientes
Pacientes com Urticária Crônica Espontânea (UCE)
Cuidados de Armazenamento
Xolair deve ser armazenado sob refrigeração entre 2 e 8 °C. Não congelar. Manter na embalagem original.
Após reconstituição, manter entre 2 e 8 °C por 8 horas ou a 30 °C por 4 horas.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Características físicas
Frasco-ampola (pó)
Pó claro, frasco-ampola de vidro incolor com tampa e selo azul (150 mg).
Ampola (solvente)
Líquido claro, ampola de vidro incolor contendo 2 mL de água para injeção.
O produto completamente reconstituído parecerá claro ou levemente opaco e pode apresentar algumas bolhas pequenas ou espuma ao redor da borda do frasco-ampola.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Mensagens de Alerta
Atenção diabéticos: contém açúcar.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.?
Todo medicamento deve ser mantido fora do alcance das crianças.
Dizeres Legais
MS – 1.0068.0983
Farm. Resp.: Flavia Regina Pegorer – CRF-SP 18.150
Importado por:
Novartis Biociências S.A.
Av. Prof. Vicente Rao, 90 – São Paulo – SP
CNPJ: 56.994.502/0001-30
Indústria Brasileira
Fabricado por: Novartis Pharma Stein AG, Stein, Suíça.
Venda sob prescrição médica.
Uso restrito a hospitais.
Esta bula foi aprovada pela ANVISA em 14/12/2015.
informações complementares
| Fabricante |
| NOVARTIS |
| Princípio ativo |
| Omalizumabe |
| Categoria do medicamento |
| Medicamentos de A-Z |