Comparamos o preço de Restasis - Emulsão Oftálmica Estéril 0,05% C 30 Flaconetes De 0,4Ml, veja o menor preço

R$ 244,03
RReferência
5
ofertasMelhores preços a partir de R$ 237,99 até R$ 311,46
Oferta patrocinada
vendido por Drogaria Nova Esperança
economize
21.65%
R$ 244,03
Mais de 47 anos de tradição, loja RA1000 e Ebit Diamante!
vendido por Pague Menos
economize
23.59%
R$ 237,99
vendido por Drogaria Soares
economize
9.73%
R$ 281,14
Enviamos para todo o Brasil. Clique e confira!
vendido por promofarma
economize
7.53%
R$ 288,00
vendido por Farmácia Indiana
R$ 311,46
Para que serve
Restasis emulsão oftálmica é indicada para aumentar a produção de lágrimas em pacientes cuja produção é supostamente suprimida devido à inflamação ocular associada à ceratoconjuntivite seca (síndrome do olho seco).
O aumento da produção de lágrimas não foi observado em pacientes recebendo medicamentos tópicos oculares anti-inflamatórios ou usando tampões para ocluir o sistema de drenagem lacrimal.
Como Restasis funciona?
Restasis é uma emulsão que contém ciclosporina. Quando administrada sistematicamente, a ciclosporina é capaz de diminuir ou impedir reações do sistema imune.
Em pacientes com produção de lágrimas diminuída devido à inflamação ocular (nos olhos) juntamente com ceratoconjuntivite seca (síndrome crônica do olho seco), acredita-se que a ciclosporina aumente a produção de anticorpos.
Tempo de Ação
O mecanismo de ação exato não é conhecido.
Contraindicação
Restasis é contraindicado para pessoas que apresentam alergia conhecida à ciclosporina ou a qualquer um dos demais componentes da fórmula do produto.
Este medicamento é contraindicado para uso por pacientes que apresentam infecção ativa nos olhos.
Como usar
- Você deve usar este medicamento exclusivamente nos olhos
- Antes de utilizar o medicamento, confira o nome no rótulo, para não haver enganos. Não utilize Restasis caso haja sinais de violação e/ou danificações no lacre do frasco.
- Inverter o flaconete algumas vezes para obter uma emulsão uniforme, opaca e branca antes de usar. Para abrir, girar totalmente a ponteira. Não puxar. Não encoste o flaconete nos olhos, nos dedos e nem em outra superfície qualquer, para evitar a contaminação do flaconete e do colírio.
- A emulsão contida no flaconete deve ser utilizada imediatamente após sua abertura. A dose usual para tratamento do olho seco é de 1 gota no(s) olho(s) afetado(s) duas vezes ao dia, com intervalo aproximado de 12 horas entre as aplicações.
- O conteúdo do flaconete deve ser descartado imediatamente depois do uso do produto. Não reutilizar.
- Não descontinue o tratamento prematuramente.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento.
Não interrompa o tratamento sem o conhecimento do seu médico.
O que devo fazer quando eu me esquecer de usar Restasis?
Você deve retornar a utilização deste medicamento assim que se lembrar seguindo normalmente os intervalos de horários entre as aplicações até o final do dia. No dia seguinte, retornar aos horários regulares.
A dose não deve ultrapassar duas gotas diárias em cada olho(s) afetado(s).
Em caso de dúvidas, procure a orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Precauções
Para não contaminar evite contato do flaconete com qualquer superfície. Não permita que o flaconete entre em contato direto com os olhos.
Restasis é um medicamento de uso exclusivamente tópico ocular.
Restasis emulsão oftálmica não foi estudado em pacientes com história de ceratite herpética (infecção da córnea devido ao vírus do herpes).
Uso durante a Gravidez e Lactação
Gravidez
Não foram realizados estudos adequados e bem controlados em mulheres durante a gestação. A emulsão oftálmica de ciclosporina 0,05% não é absorvida sistematicamente se administrada por via tópica ocular, e o uso materno não é esperado que resulte em exposição fetal à substância.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Lactação
A ciclosporina administrada por via sistêmica é excretada pelo leite humano, mas não foram realizados estudos sobre a excreção no leite humano após administração tópica. Embora as concentrações sanguíneas não sejam detectáveis após aplicação tópica, recomenda-se cautela ao administrar Restasis emulsão oftálmica a mulheres que estejam amamentando.
Uso em crianças
A segurança e eficácia de Restasis emulsão oftálmica não foi estabelecida em pacientes pediátricos.
Uso em idosos
Não foram observadas diferenças na segurança e eficácia deste medicamento entre pacientes mais jovens e idosos.
Pacientes que utilizam lentes de contato
Restasis não deve ser utilizado durante o uso de lentes de contato.
Caso você esteja utilizando lentes de contato, estas devem ser retiradas antes da aplicação de Restasis em um ou ambos os olhos, e recolocadas depois de no mínimo 15 minutos após a administração do colírio.
Pacientes que fazem uso de mais de um medicamento oftálmico
Se você for utilizar Restasis com outros colírios, aguarde um intervalo de 5 minutos entre a aplicação de cada medicamento.
Efeitos na capacidade de dirigir veículos e operar máquinas
Como acontece com qualquer tratamento ocular, se ocorrer visão turva passageira no momento do gotejamento, você deve esperar até que a visão normalize antes de conduzir ou utilizar máquinas.
Interações medicamentosas
Não foram realizados estudos específicos de Restasis para avaliar a interação com outros medicamentos.
Não são esperadas interações medicamentosas entre o uso de Restasis e medicamentos sistêmicos, uma vez que, após aplicação oftálmica, não foi observada absorção sistêmica detectável de Restasis.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde.
Reações Adversas
Como acontece com qualquer medicamento, podem ocorrer reações indesejáveis com a aplicação de Restasis emulsão oftálmica.
Reação muito comum (ocorre em mais de 10% dos pacientes que utilizam este medicamento):
Ardor ocular.
Reação comum (ocorre entre 1% e 10% dos pacientes que utilizam este medicamento):
Cefaleia (dor de cabeça), irritação nos olhos, sensação de corpo estranho nos olhos, hiperemia conjuntival (vermelhidão nos olhos), dor nos olhos, pontadas nos olhos, secreção nos olhos, fotofobia (sensibilidade anormal à luz), prurido (coceira) nos olhos, distúrbios visuais (visão turva) e olho seco.
Reação incomum (ocorre entre 0,1% e 1% dos pacientes que utilizam este medicamento):
Tontura, ceratite ulcerativa (inflamação da córnea), edema (inchaço) na pálpebra, eritema (vermelhidão) da pálpebra, aumento do lacrimejamento, náuseas e erupção cutânea.
Outras reações adversas foram reportadas após a comercialização de Restasis e podem potencialmente ocorrer são:
Inchaço dos olhos, hipersensibilidade (alergia), danos superficiais ao olho (por exemplo em casos em que a ponta do flaconete entra em contato com o olho), prurido (coceira) e sensação de queimação. Raros casos incluíram também angioedema grave, inchaço da face, inchaço da língua, edema faringeal e dispneia.
Informe ao seu médico, cirurgião-dentista ou farmacêutico o aparecimento de reações indesejáveis pelo uso do medicamento.
Informe também à empresa através do seu serviço de atendimento.
Composição
Cada ml contém:
0,5 mg de ciclosporina.
Veículo: glicerina, óleo de rícino, polissorbato 80, carbômer 1342, água purificada e hidróxido de sódio para ajuste do pH.
Superdosagem
É pouco provável que ocorra superdosagem sistêmica após administração ocular de Restasis. Devido à baixa concentração sistêmica de ciclosporina após tratamento tópico com Restasis, a probabilidade de ocorrer intoxicação sistêmica por superdosagem tópica em humanos é muito baixa.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível.
Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação Medicamentosa
Dentre os vários fármacos que interagem com a ciclosporina, estão listados abaixo aqueles cujas interações foram adequadamente documentadas e consideradas como tendo implicações clínicas.
Uso concomitante não recomendado devido à interação
Durante o tratamento com ciclosporina, a vacinação pode ser menos eficaz, o uso de vacinas vivas atenuadas deve ser evitado.
Interações a serem consideradas
Recomenda-se cautela para o uso concomitante com fármacos poupadores de potássio (por exemplo, diuréticos poupadores de potássio, inibidores da enzima conversora de angiotensina, antagonistas do receptor de angiotensina II) e fármacos contendo potássio uma vez que eles podem levar a um aumento significativo do potássio sérico.
Após a administração concomitante da ciclosporina com o lercanidipino, a AUC do lercanidipino aumentou três vezes e a AUC da ciclosporina aumentou 21%. Dessa forma, recomenda-se precaução quando da coadministração de ciclosporina com lercanidipino.
Deve-se ter cautela ao usar ciclosporina junto com metotrexato em pacientes com artrite reumatoide, devido ao risco de sinergia nefrotóxica.
Interações que diminuem os níveis de ciclosporina
Barbitúricos, carbamazepina, oxcarbazepina, fenitoína; nafcilina, sulfadimidina i.v.; rifampicina; octreotida; probucol; orlistate; Hypericum perforatum (Erva de São João); ticlopidina, sulfimpirazona, terbinafina, bosentana.
Interações que aumentam os níveis de ciclosporina
Antibióticos macrolídeos, cetoconazol, fluconazol, itraconazol, voriconazol; diltiazem, nicardipina, verapamil; metoclopramida; anticoncepcionais orais; danazol; metilprednisolona (doses elevadas), alopurinol, amiodarona, ácido cólico e derivados; inibidores de protease, imatinibe; colchicina; nefazodona.
Interações que resultam em aumento do potencial de nefrotoxicidade
Durante o uso concomitante de um fármaco que pode exibir sinergismo de nefrotoxicidade, deve-se fazer um monitoramento cuidadoso da função renal (em particular a creatinina sérica).
Se ocorrer prejuízo significativo da função renal, a dose do fármaco coadministrado deve ser reduzida ou um tratamento alternativo deve ser considerado.
Deve-se tomar cuidado ao se administrar ciclosporina juntamente com fármacos que possuem sinergismo de nefrotoxicidade como: aminoglicosídeos (incluindo gentamicina e tobramicina), anfotericina B, ciprofloxacino, vancomicina, trimetoprima (mais sulfametoxazol), anti-inflamatórios não-esteroidais (incluindo diclofenaco, naproxeno, sulindaco), melfalana, antagonistas de receptores histamínicos H2 (por exemplo, cimetidina, ranitidina), metotrexato
O uso concomitante com tacrolimo deve ser evitado devido ao aumento potencial de nefrotoxicidade.
A administração concomitante de diclofenaco com ciclosporina resulta em aumento significante da biodisponibilidade do diclofenaco, com a possível consequência de diminuição reversível da função renal.
O aumento da biodisponibilidade do diclofenaco parece estar mais relacionado com uma redução no seu elevado efeito de primeira passagem. Portanto, se for administrado juntamente com a ciclosporina um anti-inflamatório não-esteroidal com reduzido efeito de primeira passagem (como o ácido acetilsalicílico), este aumento da biodisponibilidade não é esperado.
Fármacos anti-inflamatórios não-esteroidais que possuem efeito de primeira passagem pronunciado (como o diclofenaco) devem ser administrados em doses menores do que aquelas que seriam utilizadas em pacientes que não estão recebendo ciclosporina.
Em receptores de enxertos foram relatados casos isolados de considerável, porém reversível diminuição da função renal (com aumento correspondente na creatinina sérica) após administração concomitante de derivados de ácido fíbrico (por exemplo, bezafibrato, fenofibrato). A função renal deve, entretanto, ser cuidadosamente monitorada nestes pacientes.
Nos eventos de significativa diminuição da função renal a comedicação deve ser retirada.
Interações que resultam no aumento da taxa de hiperplasia gengival
A administração concomitante de ciclosporina com nifedipino pode resultar em aumento da frequência de hiperplasia gengival comparada com a administração isolada de ciclosporina.
O uso concomitante de nifedipino deve ser evitado em pacientes em que se observou desenvolvimento de hiperplasia gengival como efeito adverso da ciclosporina
Interações que resultam no aumento de outros fármacos
A ciclosporina também é um inibidor da CYP3A4 e do transportador de efluxo multifármaco glicoproteína-P, e pode aumentar os níveis plasmáticos das comedicações que são substratos desta enzima e/ou transportador.
A ciclosporina pode reduzir o clearance (depuração) da digoxina, colchicina, prednisolona, de inibidores da HMG-CoA redutase (estatinas) e do etoposídeo.
Em muitos pacientes que tomam digoxina, foi observada toxicidade digitálica severa após poucos dias do início da ciclosporina.
Também há relatos que a ciclosporina aumenta os efeitos tóxicos da colchicina tais como miopatia e neuropatia, especialmente em pacientes com disfunção renal. Se a digoxina ou colchicina forem usadas concomitantemente com ciclosporina é necessária observação clínica para possibilitar a detecção antecipada de manifestações tóxicas de digoxina ou colchicina, seguida pela redução da dosagem ou pela sua retirada.
Foram relatados casos na literatura e pós-comercialização de miotoxicidade, incluindo dor muscular e fraqueza, miosite e rabdomiólise, com a administração concomitante de ciclosporina com lovastatina, sinvastatina, atorvastatina, pravastatina e raramente fluvastatina.
Quando concorrentemente administrado com ciclosporina, a dose destas estatinas deve ser reduzida de acordo com as recomendações na bula. A terapia com estatina necessita ser temporariamente suspensa ou descontinuada em pacientes com sinais e sintomas de miopatia ou daqueles com fatores de risco de prédisposição para dano renal severo, incluindo falência renal, secundariamente para rabdomiólise.
Se digoxina, colchicina ou os inibidores da enzima HMG-CoA redutase (estatinas) forem administrados concomitantemente com ciclosporina, é necessário a observação clínica rigorosa a fim de se permitir a detecção precoce de manifestações tóxicas dos fármacos, seguida de redução de sua dose ou sua retirada.
Elevações na creatinina sérica foram observadas em estudos usando everolimo e sirolimo em combinação com ciclosporina dose-completa para microemulsão. Este efeito é frequentemente reversível com a redução da dose de ciclosporina.
O everolimo e sirolimo têm pouca influência na farmacocinética da ciclosporina. A coadministração de ciclosporina significativamente aumenta os níveis no sangue de everolimo e sirolimo.
A ciclosporina pode aumentar as concentrações plasmáticas de repaglinida e, desta forma, aumentar o risco de hipoglicemia.
A coadministração de bosentana e ciclosporina em voluntários sadios resultou em um aumento de aproximadamente 2 vezes na exposição da bosentana e um decréscimo de 35% na exposição da ciclosporina.
Seguindo a administração concomitante de ciclosporina e alisquireno, o Cmax do alisquireno foi aumentado aproximadamente 2,5 vezes e a AUC em aproximadamente 5 vezes. No entanto, o perfil farmacocinético da ciclosporina não teve alteração significativa.
A administração concomitante de dabigatran e ciclosporina leva ao aumento do nível plasmático de dabigatran devido à inibição da atividade da PgP de ciclosporina. O dabigatran tem um índice terapêutico estreito e um aumento no nível plasmático que pode ser associado com um risco aumentado de hemorragia.
A administração de doses múltiplas de ambrisentana e ciclosporina em voluntários sadios resultou em um aumento de aproximadamente 2 vezes na exposição de ambrisentana, enquanto a exposição de ciclosporina aumentou levemente (aproximadamente 10%).
Um aumento significativo na exposição de antibióticos antraciclínicos (por exemplo, doxorrubicina, mitoxantrona, daunorrubicina) foi observado em pacientes oncológicos com coadministração intravenosa de antibiótico antraciclínico e doses elevadas de ciclosporina.
Interações a serem consideradas que aumentam ou diminuem os níveis de ciclosporina
Vários agentes são conhecidos por aumentar ou diminuir os níveis plasmáticos ou sanguíneos da ciclosporina, geralmente por inibição ou indução de enzimas envolvidas no metabolismo da ciclosporina, especialmente da CYP3A4.
Se o uso concomitante de fármacos que interagem com a ciclosporina não pode ser evitado, as seguintes recomendações básicas devem ser observadas:
Em pacientes transplantados
Medição frequente dos níveis de ciclosporina e, se necessário, ajuste de dose da ciclosporina, particularmente durante a introdução ou retirada do fármaco coadministrado;
Em pacientes não-transplantados
O valor do monitoramento do nível sanguíneo da ciclosporina é questionável, já que nestes pacientes a relação entre o nível sanguíneo e os efeitos clínicos não estão bem estabelecidos. Se fármacos que aumentam os níveis de ciclosporina foram administrados concomitantemente, a avaliação frequente da função renal e o monitoramento cuidadoso de efeitos adversos relacionados à ciclosporina pode ser mais apropriada do que a avaliação dos níveis sanguíneos.
Interação Alimentícia
Foi relatado que a ingestão concomitante de suco de toranja (grapefruit) aumenta a biodisponibilidade da ciclosporina.
Ação da Substância
Resultados da eficácia
Transplante de órgãos sólidos
Foi demonstrada a eficácia em 13 estudos globais que avaliaram o sucesso na taxa de transplantes utilizando ciclosporina em comparação a outros agentes imunossupressores. Foram realizados estudos clínicos em diversas regiões (Europa, Austrália e América do Norte).
Alguns destes estudos incluíram uma avaliação de diferentes órgãos sólidos, incluindo transplante alogênico de rins, fígado, coração, coração-pulmão combinados, pulmão ou de pâncreas. Nos estudos clínicos realizados, a dose de ciclosporina utilizada nos pacientes submetidos a transplante variou de 10 a 25 mg/kg ao dia como dose inicial do tratamento e variou de 6 a 8 mg/kg ao dia como dose de manutenção.
Os estudos clínicos são apresentados nas Tabelas 1 a 4 a seguir.
Transplante de rins e pâncreas
A Tabela 1 apresenta os estudos clínicos realizados principalmente em pacientes submetidos a transplante renal e a Tabela 2 apresenta os estudos clínicos realizados apenas em pacientes submetidos a transplante renal. A Tabela 1 também inclui os pacientes submetidos a transplante de pâncreas.
Os estudos inclusos nestas tabelas confirmam que a ciclosporina administrada em combinação com esteroides é um tratamento eficaz no transplante renal. A sobrevida em um ano após enxerto aumentou significativamente nos pacientes tratados com ciclosporina em comparação à terapia controle.
Tabela 1. Transplante de órgãos sólidos – Estudos clínicos europeus e estudo clínico australiano
| Número do Estudo/País | Características do Estudo | Órgão (N) |
| Estudo Nº 1 Cambridge, RU | Centro único CsA vs. Histórico AZA+CS | Rim (63) Fígado (7) Pâncreas (10) Incluindo Rim/Pâncreas (7) Rim/Fígado (1) Pâncreas/Fígado (1) |
| Estudo Nº 2 Austrália |
Centro único, randomizado | Rim (29 total; 14 Ciclosporina) |
| Estudo Nº 3 Europeu Estudo multicêntrico | Multicêntrico, randomizado CsA vs AZA+Pred | Rim (232 total; 117 Ciclosporina) |
| Estudo Nº 4 Suécia | Centro único; CsA (4 pacientes) CsA + Pred (16) vs. Controle histórico | Rim (20) |
| Estudo Nº 5 Finlândia |
Multicêntrico | Rim (9) (32) (32) |
RU: Reino Unido; CsA: ciclosporina; AZA: azatioprina; CS: corticosteroides; ALG: globulina antilinfócito; Pred: prednisona; MP: metilprednisolona; IV: intravenoso; N: número de pacientes.
Tabela 2. Transplante de órgãos sólidos – Estudos clínicos norte-americanos
| Número do Estudo País | Características do Estudo | Órgão (N) |
| Estudo Nº 2 EUA | Grupo I: CsAa + TDD Grupo II: CsAb Grupo III: CsAc Todos os pacientes receberam CS | Rim Grupo I: 12 Grupo II: 20 Grupo III: 34 |
| Estudo Nº 5 EUA | CsA +baixa dose de Pred vs. AZA+ ATG | Rim (98 total; 47 CsA) |
| Estudo Nº 7 EUA | CsA + CS+ diuréticos vs. AZA+ CS+ diuréticos | Rim (27 total; 14 CsA) |
| Estudo Nº 15 EUA | Aberto, randomizado CsA+Pred vs. AZA+Pred | Rim (41 total; 21 CsA) |
| Canadense, Multicêntrico | Randomizado, CsA vs. AZA + CS | Rim (209 ; 103 CsA) |
TDD: drenagem do ducto torácico; CsA: ciclosporina; CS: corticosteroides; Pred: prednisona; ATG: globulina antilinfócito; AZA: azatioprina; a. CsA: administrada em dose única no dia do transplante e posteriormenteb. CsA: administrada 2-30 dias antes do transplante, sem TDDc. CsA: administrada em dose única no dia do transplante e posteriormente sem TDD.
Transplante hepático
No transplante hepático (vide Tabela 3), os estudos clínicos demonstraram que a taxa de sobrevida em um ano dos pacientes foi mais alta no grupo que recebeu ciclosporina do que nos controles históricos que estavam sob regimes imunossupressores prévios.
A maior parte dos treze óbitos foi atribuída a complicações cirúrgicas, a infecções agudas (que geralmente ocorreram imediatamente após o transplante e que, possivelmente, foram causadas por procedimentos em órgãos e para preservação) ou a recidiva da doença inicial.
Os episódios de rejeição aguda geralmente foram controlados ao se aumentar a administração de esteroides. Por outro lado, foram observados episódios de nefrotoxicidade que foram resolvidos com redução da dose de ciclosporina.
Os estudos clínicos demonstraram que a terapia com ciclosporina e esteroides apresenta uma vantagem considerável em comparação à terapia padrão com azitromicina e esteroides.
Tabela 3. Transplante de órgãos sólidos – Estudos hepáticos

CsA: Ciclosporina / CS: Corticosteroides / TDD: Drenagem de ducto torácico.
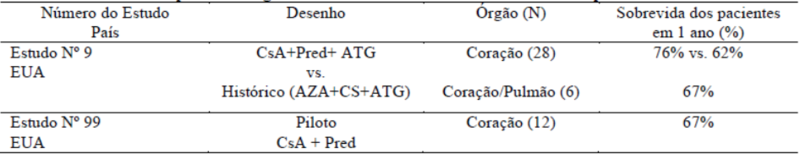
Transplante cardíaco e cardíaco-pulmonar
No transplante cardíaco, os estudos clínicos demonstraram que as taxas de sobrevida em um ano e 18 meses dos pacientes foram significativamente mais altas nos pacientes tratados com ciclosporina do que nos pacientes do grupo controle. Dez dos 28 pacientes incluídos em transplante cardíaco não apresentaram episódios de rejeição após o transplante.
No transplante cardíaco-pulmonar, a taxa de sobrevida em um ano foi de 67% nos pacientes tratados com ciclosporina. Tanto em transplantes cardíacos quanto em transplantes cardíaco-pulmonares, os episódios de hepatotoxicidade e nefrotoxicidade suspeitas foram controlados ao se reduzir a dose de ciclosporina. Infecções pulmonares sérias foram observadas e a maioria delas foi tratada com sucesso.
Os resultados dos estudos clínicos realizados nos pacientes submetidos a transplante cardíaco ou cardíaco-pulmonar estão resumidos na Tabela 4 a seguir:
Tabela 4. Transplante de órgãos sólidos - Estudos Cardíacos e Cardíacos-pulmonares
Transplante de medula óssea
A eficácia de ciclosporina (na forma farmacêutica convencional) em receptores de transplante de medula óssea (BMT) foi demonstrada em oito estudos realizados na Europa e nos EUA com um total de 227 pacientes.
Foram realizados sete estudos para a prevenção da doença do enxerto versus hospedeiro (GVHD) e um estudo para o tratamento da GVHD aguda. Cinco centros europeus (UE 1-5) e um centro dos EUA (EUA n° 6) realizaram estudos não randomizados “abertos” para a prevenção da GVHD.
Um estudo randomizado (EUA n° 3) foi realizado para a prevenção da GVHD e um estudo randomizado (EUA N° 11) foi realizado para o tratamento da GVHD aguda.
Seis pacientes no estudo (EUA n° 6) receberam ciclosporina na tentativa de reverter a GVHD aguda e grave estabelecida (Grau III-IV). Estes pacientes não receberam tratamento anterior com ciclosporina e a GVHD foi resistente a outras terapias.
Os resultados destes estudos foram comparados a estudos da terapia com metotrexato (MTX) na prevenção da GVHD (controles históricos nos estudos abertos) e a um estudo da terapia com esteroides no tratamento da GVHD. Estes estudos continham 227 pacientes: 204 pacientes haviam recebido BMT e haviam recebido tratamento para profilaxia da GVHD e 23 pacientes haviam recebido tratamento para a GVHD estabelecida. No total, havia 20 pacientes com incompatibilidade ao HLA nestes estudos.
A dose de ciclosporina variou nos diferentes estudos. A dose habitual para a prevenção da GVHD era de 12,5 mg/kg/dia. Contudo, diversos centros europeus iniciaram com uma dose mais alta (20-25 mg/kg/dia) durante os primeiros dias e, então, reduziram a dose gradualmente para 12,5 mg/kg/dia. A maioria dos centros manteve a dose inalterada e a reduziram após vários meses, geralmente descontinuando a dose após 4-6 meses.
A dose de ciclosporina utilizada para o tratamento da GVHD foi de aproximadamente 15 mg/kg/dia. Esta dose foi reduzida gradualmente com o passar do tempo e foi descontinuada em cerca de 6 meses. Na maioria das vezes, ciclosporina era administrada uma ou duas vezes ao dia, porém, em um centro, ela era administrada três vezes ao dia. Na maioria dos estudos, caso a formulação I.V. da ciclosporina fosse utilizada, ela era administrada a cerca de 1/3 da dose oral.
Os resultados de eficácia nos estudos europeus demonstraram uma redução da gravidade e talvez da frequência da GVHD, com uma sobrevida de um ano para todos os pacientes que receberam ciclosporina e com enxertos compatíveis em aproximadamente 70% dos casos.
Para os controles históricos tratados com MTX, o número foi de apenas 52% para uma sobrevida de um ano. Ocorreu óbito associado à GVHD em apenas 10/132 pacientes (8%), uma taxa muito menor do que a observada anteriormente com uso de MTX em enxertos compatíveis (fatal em >25% dos casos).
Os resultados de eficácia dos estudos realizados nos EUA corroboram os resultados de eficácia de estudos europeus e demonstram que a ciclosporina é, no mínimo, tão eficaz quanto e provavelmente superior à terapia com MTX na prevenção da GVHD no BMT, com um tempo significativamente mais rápido para o enxerto e um risco relativo de 50% de desenvolvimento da GVHD maior do que Grau II ou III (p=N.S.). O estudo EUA n° 6 também demonstrou que a ciclosporina reverteu a GVHD aguda e severa (Grau III-IV) estabelecida nos pacientes que não receberam tratamento anterior com ciclosporina e resistentes a outras terapias.
Indicações não relacionadas a transplante
Uveíte endógena, incluindo uveíte de Behçet
A eficácia da ciclosporina foi demonstrada em 11 estudos não controlados abertos da Europa, dos EUA, do Japão, da África e da Ásia, incluindo 242 pacientes com uveíte endógena, sendo que na maioria destes pacientes a terapia convencional falhou ou causou eventos adversos inaceitáveis.
Em 4 estudos controlados mascarados de Israel, do Japão, dos Países Baixos e dos EUA, 202 pacientes foram designados randomicamente para receber ciclosporina (97 pacientes) ou terapia convencional – prednisolona, clorambucil, colchicina – (92 pacientes) ou placebo (13 pacientes).
Dos 339 pacientes tratados com ciclosporina, 161 receberam o diagnóstico de uveíte de Behçet e os outros 178 receberam, predominantemente, o diagnóstico de uveíte intermediária ou posterior de etiologia não infecciosa.
Havia 201 pacientes do sexo masculino e 138 do sexo feminino; a idade média foi de 35,8 anos. A maioria dos pacientes que recebiam ciclosporina receberam uma dose de reforço inicial de 5 a 10 mg/kg/dia, seguida por uma redução da dose de acordo com a atividade inflamatória ocular e com a tolerabilidade. Melhora da acuidade visual desde o basal foi o desfecho primário utilizado mais comumente no programa clínico, e a incidência de ataques oculares foi utilizada para a uveíte de Behçet.
Mais de 60% dos pacientes tratados com ciclosporina apresentaram melhora na acuidade visual desde o basal em comparação à melhora em 3 e 6 meses após o início da terapia com ciclosporina.
O fator limitante inicial para a melhora na acuidade na maioria dos outros 40% foram as alterações irreversíveis que ocorreram durante o processo da doença antes do início da terapia com ciclosporina.
A incidência de ataques oculares em pacientes com uveíte de Behçet foi significativamente reduzida (p=0,001) nos pacientes tratados com ciclosporina em comparação aos pacientes tratados com colchicina.
Síndrome nefrótica
A eficácia de ciclosporina (na forma farmacêutica convencional) foi demonstrada em quatro estudos randomizados controlados e 5 estudos não controlados. Os resultados clínicos desses nove estudos clínicos foram analisados ao se reunir dados de todos os estudos (controlados e não controlados).
Dois estudos multicêntricos, duplo-cegos e controlados por placebo (9515 e 9516) e um estudo multicêntrico para comparar a ciclosporina e a ciclofosfamida em pacientes com resistência a esteroides (9508) tiveram de ser interrompidos prematuramente devido à falta de pacientes adequados que concordaram em receber placebo ou um agente citostático.
Uma coleta retrospectiva dos dados de pacientes tratados com ciclosporina foi realizada em um estudo intitulado OL 03. Os pacientes adultos e pediátricos incluídos nestes estudos eram predominantemente resistentes ou dependentes de esteroides ou pacientes com sinais de toxicidade de esteroides que necessitavam de tratamento alternativo.
Os estudos controlados incluíram 47 pacientes dos quais 43 eram crianças (definidos como pacientes com até 16 anos de idade). Estes pacientes apresentavam glomeruloesclerose segmentar focal (GESF), nefropatia com alteração mínima (LGM) e glomerulonefrite membranosa (GM) e eram dependentes ou resistentes a esteroides. Outros 24 pacientes adultos com nefropatia por IgA (uma entidade que pode apresentar síndrome nefrótica, muito comum em pacientes de origem asiática) também foram estudados.
Os estudos compararam a ciclosporina com ciclofosfamida (OL9511), clorambucil (OL9505), placebo (OL9509) ou “nenhum tratamento” ou tratamento paliativo (OL9510).
Os estudos não controlados estudaram 361 pacientes adultos e 178 pacientes pediátricos (1-17 anos) com GESF, LGM e síndrome nefrótica com GM e que eram dependentes ou resistentes a esteroides.
Outros 9 pacientes adultos e 27 pediátricos com formas de GESF que apresentavam recidiva com frequência e síndrome nefrótica com LGM foram estudados.
Dos 9 estudos descritos neste documento, sete incluíram pacientes pediátricos com 1 a 17 anos de idade. Um estudo controlado (OL9505) e um estudo não controlado (OL9504) foram realizados exclusivamente na população pediátrica.
No total, 398 crianças (319 tratadas com ciclosporina) foram incluídas nestes estudos.
Os resultados de eficácia e segurança dos estudos realizados com crianças foram semelhantes aos da população adulta.
A maioria dos pacientes dependentes de esteroides atingiu remissão completa. A eliminação da ciclosporina é afetada pela idade dos pacientes. Os pacientes pediátricos eliminam o medicamento mais rapidamente do que os adultos com base no peso corporal.
Assim, os pacientes pediátricos precisam de doses mais altas de ciclosporina por quilograma do peso corporal para atingir concentrações séricas do medicamento semelhantes às observadas em adultos.
Artrite reumatoide
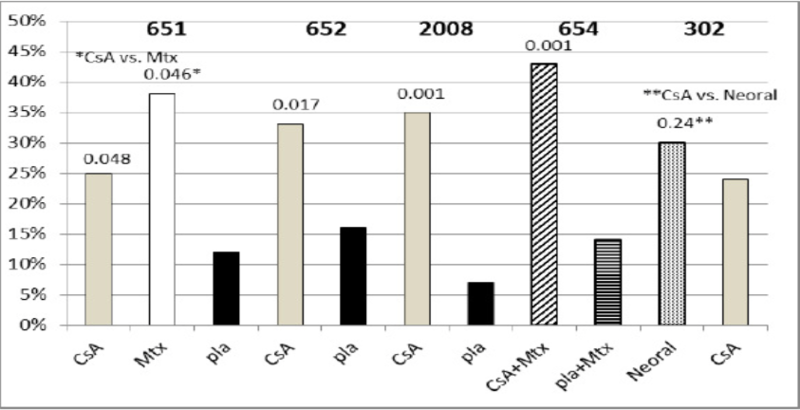
A eficácia de ciclosporina no tratamento da artrite reumatoide grave foi avaliada em 5 estudos clínicos que envolveram 728 pacientes tratados com ciclosporina e 273 pacientes tratados com placebo.
Um resumo dos resultados é apresentado para as taxas de “responsivos” por grupo de tratamento, sendo que um responsivo é definido como um paciente que concluiu o estudo com uma melhora de 20% nas contagens de articulações sensíveis e inchadas e uma melhora de 20% em 2 de 4 taxas globais do investigador, globais do paciente, incapacidade e velocidade de hemossedimentação (VHS) para os Estudos 651 e 652 e em 3 de 5 taxas globais do investigador, global do paciente, incapacidade, escala analógica visual de dor e VHS para os Estudos 2008, 654 e 302.
O estudo 651 incluiu 264 pacientes com artrite reumatoide ativa com, no mínimo, 20 articulações envolvidas, os quais apresentaram falha em pelo menos um importante medicamento para AR, utilizando uma randomização em uma proporção 3:3:2 para um dos três grupos a seguir:
- Ciclosporina a uma dose de 2,5-5 mg/kg/dia;
- Metotrexato a uma dose de 7,5-15 mg/semana;
- Placebo.
A duração do tratamento era de 24 semanas.
A dose média de ciclosporina na última visita era de 3,1 mg/kg/dia.
O estudo 652 incluiu 250 pacientes com AR ativa com > 6 articulações dolorosas ou inchadas ativas e que apresentaram falha em pelo menos um importante medicamento para AR. Os pacientes foram randomizados utilizando uma randomização em uma proporção 3:3:2 para 1 dos 3 braços de tratamento a seguir:
- 1,5-5 mg/kg/dia de ciclosporina;
- 2,5-5 mg/kg/dia de ciclosporina;
- Placebo.
A duração do tratamento era de 16 semanas. A dose média de ciclosporina para o grupo 2 na última visita era de 2,92 mg/kg/dia.
O estudo 2008 incluiu 144 pacientes com AR ativa com > 6 articulações ativas e que apresentaram falha no tratamento com aspirina e sais de ouro ou penicilina. Os pacientes foram randomizados para 1 de 2 grupos de tratamento:
- 2,5-5 mg/kg/dia de ciclosporina com ajustes após o primeiro mês para atingir um nível alvo mínimo;
- Placebo.
A duração do tratamento era de 24 semanas. A dose média de ciclosporina na última visita era de 3,63 mg/kg/dia.
O estudo 654 incluiu 148 pacientes que permaneceram com contagens de 6 ou mais articulações ativas apesar do tratamento com as doses máximas toleradas de metotrexato por, no mínimo, três meses.
Os pacientes continuaram a utilizar sua dose atual de metotrexato e foram randomizados para receber, adicionalmente, um dos medicamentos a seguir:
- 2,5 mg/kg/dia de ciclosporina com aumentos de dose de 0,5 mg/kg/dia nas semanas 2 e 4, caso não houvesse evidência de toxicidade, e aumentos de 0,5 mg/kg/dia nas semanas 8 e 16 caso ocorresse uma redução < 30% na contagem de articulações ativas sem nenhuma toxicidade significativa; reduções da dose podiam ser realizadas a qualquer momento no caso de toxicidade;
- Placebo.
A duração do tratamento era de 24 semanas. A dose média de ciclosporina na última visita era de 2,8 mg/kg/dia (faixa: 1.3-4.1).
O estudo 302 incluiu 299 pacientes com AR ativa severa, sendo que 99% destes apresentavam intolerância ou não eram responsivos a no mínimo um importante medicamento para AR utilizado anteriormente.
Os pacientes foram randomizados para 1 de 2 grupos de tratamento:
- Ciclosporina microemulsão;
- Ciclosporina (na forma farmacêutica convencional).
Os dois grupos iniciavam com uma dose de 2,5 mg/kg/dia, a qual era aumentada após 4 semanas no caso de ineficácia, sendo que os aumentos eram de 0,5 mg/kg/dia a no máximo 5 mg/kg/dia. A dose era reduzida a qualquer momento no caso de toxicidade. A duração do tratamento era de 24 semanas. A dose média de ciclosporina na última visita era de 2,91 mg/kg/dia (faixa: 0,72-5,17) para ciclosporina microemulsão e 3,27 mg/kg/dia (faixa: 0,73-5,68) para ciclosporina (na forma farmacêutica convencional).
Figura 1. Eficácia da ciclosporina no tratamento de artrite reumatoide severa em 5 estudos clínicos (651, 652, 2008, 654 e 302)
Psoríase
A eficácia da ciclosporina foi demonstrada em 1.270 pacientes com psoríase grave em 13 estudos clínicos. Três principais estudos duplo-cegos, controlados por placebo e que incluíram um total de 296 pacientes, dentre os quais 199 foram tratados com ciclosporina e 97 com placebo, foram realizados durante um período de tratamento de 12-16 semanas (Estudos US299, US501 e US502); estudos menores controlados por placebo (Estudos OL8002, OL8003, OL8006 e CyA40) e que incluíram 105 pacientes, dos quais 53 foram tratados com ciclosporina e 52 com placebo, apoiaram o uso a curto prazo.
Dois estudos amplos (Estudos OL8013 e OL8014) e que incluíram 405 pacientes, dos quais 192 foram tratados com ciclosporina e 38 com etretinato, forneceram informações sobre a eficácia, a segurança e a tolerabilidade a longo prazo de diferentes doses de ciclosporina.
As duas formulações de ciclosporina foram comparadas diretamente em um estudo multicêntrico, randomizado e duplo-cego que incluiu 309 pacientes (Estudo OLP302), corroborado por um estudo menor de PK que incluiu 39 pacientes (estudo N101) e por um estudo de investigação (Estudo OL8095) em que a formulação de microemulsão foi administrada intermitentemente a 41 pacientes.
Os pacientes tratados no programa clínico eram adultos com psoríase grave, nos quais a terapia convencional foi ineficaz ou inapropriada. Diversas medições primárias de eficácia foram utilizadas nos estudos clínicos, ou seja, as pontuações de avaliação geral e global avaliadas pelos investigadores, o tempo até a recidiva, a avaliação da área de superfície corporal (ASC), a avaliação do índice da área e severidade da psoríase (pontuação PASI).
Os resultados de uma análise agrupada dos 3 principais estudos duplo-cegos e controlados por placebo (Estudos US299, US501 e US502) apresentaram uma redução de, no mínimo, 75% no PASI numa faixa que variou de 76% dos pacientes tratados com uma dose inicial de 3 mg/kg/dia a 100% dos pacientes tratados com uma dose inicial de 7,5 mg/kg/dia, sendo que 83% dos pacientes foram tratados com 5 mg/kg/dia.
O maior percentual de pacientes no grupo que recebeu placebo foi de 4%. Os resultados da análise agrupada de outros estudos (Estudos 8002, 8003, 8006, CyA-40, 8013 e 8014) revelaram uma redução de, no mínimo, 75% no PASI em 55% dos pacientes tratados com uma dose inicial de 2,5 mg/kg/dia a 87% dos pacientes tratados com uma dose inicial de 5 mg/kg/dia.
Foi observada uma redução de, no mínimo, 75% no PASI em 72% dos 152 pacientes tratados com ciclosporina para microemulsão e em 62% dos 156 pacientes tratados com ciclosporina (Estudo OLP302); nos dois braços de tratamento, a dose inicial foi de 2,5 mg/kg/dia.
Dermatite atópica
A eficácia da ciclosporina sobre a dermatite atópica grave foi demonstrada em 2 estudos cruzados prospectivos, duplocegos e controlados por placebo realizados durante um período de tratamento de 8 semanas (SIM 79 e SIM 80) e em um estudo duplo-cego e controlado por placebo durante um período de tratamento de 6 semanas (SIM 24).
Uma dose de 5 mg/kg ao dia foi utilizada no decorrer destes três estudos. Três outros estudos abertos não controlados (SIM AD01, SIM AD02 e OL10085), um estudo randomizado e controlado para a determinação da dose (SIM AD 5-4-3/3-4-5) e um estudo de centro único (SIM SF04) foram realizados para examinar as taxas de recidiva após a retirada da ciclosporina ou para avaliar os efeitos da terapia a longo prazo e de diferente estratégias de dose.
Em um destes estudos (SIM SF04) foram administrados 5 mg/kg ao dia do medicamento durante 6 semanas e, então, as taxas de recidiva foram observadas durante outras 6 semanas; os pacientes que apresentaram recidiva receberam um segundo ciclo de ciclosporina e foram monitorados novamente quanto à ocorrência de recidiva.
Nos estudos a longo prazo (SIM AD02, OL 10085, SIM AD 5-4-3/3-4- 5), a dose de ciclosporina foi ajustada de acordo com a resposta e com os efeitos colaterais. Em diversos estudos abertos, os pacientes iniciaram sob uma baixa dose de ciclosporina (2,5-3,0 mg/kg/dia), a qual era ajustada caso fosse necessário.
Em todos os estudos clínicos, com exceção do OL 10901 em que foi utilizada microemulsão de ciclosporina, foi utilizada a formulação de ciclosporina baseada em óleo.
No total, 376 pacientes foram incluídos nestes 9 estudos; 296 pacientes foram tratados com ciclosporina, 23 com placebo e 57 com ciclosporina e placebo nos dois estudos cruzados (SIM 79, SIM 80).
No total, 259 pacientes foram tratados com ciclosporina a curto prazo (89 durante 6 semanas e 170 durante 8 semanas); 117 pacientes foram envolvidos em estudos a longo prazo, sendo que 100 deles foram tratados por no mínimo 12 meses. Os pacientes tratados no programa clínico eram adultos com dermatite atópica grave e em quem a terapia convencional foi ineficaz ou inapropriada.
Nos estudos controlados e na maioria dos estudos abertos, as medições primárias de eficácia foram sobre a área de envolvimento cutâneo e a gravidade da doença cutânea. Outras medições incluíram as pontuações de coceira e de perda de sono.
Os resultados dos estudos controlados por placebo (SIM 79, SIM 80 e SIM 24) demonstraram que a ciclosporina foi eficaz na maioria dos pacientes com dermatite atópica grave; apenas 5 dos 80 pacientes tratados com ciclosporina nestes estudos não responderam à terapia.
Os resultados dos estudos a longo prazo mostraram que a eficácia podia ser mantida utilizando-se doses menores que 5 mg/kg ao dia ao longo destes estudos, embora seja difícil avaliar os efeitos da evolução natural da doença nos resultados a longo prazo.
No estudo SIM SF04, 43% e 52% dos pacientes apresentaram recidiva 2 semanas após a interrupção do primeiro e do segundo ciclo da terapia com ciclosporina, respectivamente; a taxa de recidiva aumentou para 71 e 87% após 6 semanas, respectivamente.
Características Farmacológicas
Princípio ativo
A ciclosporina, baseada no princípio de microemulsão, reduz a variabilidade dos parâmetros farmacocinéticos e proporciona linearidade entre a dose e a exposição à ciclosporina, com um perfil de absorção mais consistente e menor influência da ingestão concomitante de alimentos.
A formulação é um pré-concentrado para microemulsão com a qual se observou, em estudos farmacocinéticos e clínicos, que a correlação entre a concentração e a exposição à ciclosporina é muito maior quando a ciclosporina é administrada como ciclosporina para microemulsão do que como ciclosporina (na forma farmacêutica convencional).
A formação da microemulsão ocorre na presença de água, tanto na forma de bebida como na de fluido gástrico.
Grupo farmacoterapêutico: agente imunossupressor, inibidor de calcineurina (código ATC L04A D01).
Mecanismo de ação / farmacodinâmica
A ciclosporina (também conhecida como ciclosporina A) é um polipeptídio cíclico que contém 11 aminoácidos. É um potente agente imunossupressor que prolonga a sobrevida de transplantes alogênicos de pele, coração, rins, pâncreas, medula óssea, intestino delgado ou pulmão em animais.
Diversos estudos sugerem que a ciclosporina inibe o desenvolvimento das reações de células mediadoras, incluindo-se imunidade a aloenxertos, hipersensibilidade cutânea tardia, encefalomielite alérgica experimental, artrite por adjuvante de Freund, doença enxerto-versus-hospedeiro (GVHD) e também produção de anticorpos dependentes de célula T.
No nível celular, inibe a produção e a liberação de linfocinas, inclusive a interleucina-2 (fator de crescimento de células T, TCGF). Ao que parece, a ciclosporina bloqueia os linfócitos durante a fase G0 ou fase G1 do ciclo celular e inibe a liberação de linfocinas, desencadeada por antígenos, pelas células T ativadas.
Todas as evidências sugerem que a ciclosporina atua especificamente e de maneira reversível nos linfócitos. Ao contrário dos agentes citostáticos, a ciclosporina não deprime a hematopoiese e não tem efeito algum sobre a função das células fagocitárias. Os pacientes tratados com ciclospórina são menos propensos a infecções do que aqueles tratados com outro tipo de terapia imunossupressora.
Realizaram-se com sucesso, no ser humano, transplantes de medula óssea e de órgãos sólidos, usando-se ciclosporina para prevenir e tratar a rejeição e a GVHD. A ciclosporina foi usada com sucesso em ambos os casos de vírus positivo ou negativo para a Hepatite C em receptores de transplante de fígado.
Foram também constatados efeitos benéficos da terapia com ciclosporina em diversas afecções consideradas ou reconhecidas como de origem autoimune.
Farmacocinética
Quando se administra ciclosporina, proporciona-se melhoria da linearidade da dose na exposição à ciclosporina (AUCB), perfil de absorção mais consistente e menos influência da ingestão concomitante de alimentos e do ritmo diurno, em comparação com ciclosporina (na forma farmacêutica convencional).
Essas propriedades combinadas produziram variabilidade intrapaciente mais baixa na farmacocinética da ciclosporina e correlação mais forte entre a concentração mínima e a exposição total (AUCB).
Como consequência dessas vantagens adicionais, o horário de administração da ciclosporina e precisa mais levar em consideração o horário das refeições. Além disso, ciclosporina para microemulsão produz uma exposição mais uniforme à ciclosporina durante todo o dia e de um dia para outro, no esquema de manutenção.
Os dados disponíveis indicam que após uma conversão 1:1 de ciclosporina (na forma farmacêutica convencional) para ciclosporina para microemulsão, as concentrações mínimas no sangue total são comparáveis, permanecendo, portanto, na margem do nível terapêutico mínimo desejado.
Em comparação com ciclosporina na forma farmacêutica convencional (com um pico de concentração plasmática atingido entre 1 a 6 horas), ciclosporina para microemulsão é absorvida mais rapidamente (produzindo um tmáx médio 1 hora antes e uma Cmáx média 59% mais elevada) e apresenta, em média, biodisponibilidade 29% superior.
A ciclosporina distribui-se amplamente fora do volume sanguíneo. No sangue, 33% a 47% estão presentes no plasma, 4% a 9% nos linfócitos, 5% a 12% nos granulócitos e 41% a 58% nos eritrócitos. No plasma, aproximadamente 90% estão ligados às proteínas, principalmente lipoproteínas.
A ciclosporina é extensivamente biotransformada em aproximadamente 15 metabólitos, não havendo uma via metabólica principal única. A eliminação é principalmente biliar e somente 6% da dose oral é excretada na urina; somente 0,1% é excretada na urina, na forma não alterada.
Existe uma alta variabilidade de dados registrados sobre a meia-vida terminal da ciclosporina, conforme o método de ensaio aplicado e a população-alvo. A meia-vida terminal oscilou entre 6,3 horas em voluntários sadios e 20,4 horas em pacientes com doença hepática grave.
População especial
Insuficiência renal
Em um estudo realizado em pacientes com insuficiência renal terminal, após uma infusão intravenosa de 3,5 mg/kg durante 4 horas, resultou em nível sanguíneo médio do pico de 1.800 ng/mL (intervalo de 1.536 a 2.331 ng/mL). O volume médio de distribuição (Vdss) foi de 3,49 L/kg e o clearance (depuração) sistêmico foi 0,369 L/h/kg.
Este clearance sistêmico (0,369 L/h/kg) foi de aproximadamente dois terços do clearance sistêmico médio (0,56 L/h/kg) em pacientes com rins funcionando normalmente. A insuficiência renal não teve efeito significativo na eliminação da ciclosporina.
Insuficiência hepática
Em um estudo realizado em pacientes com doença grave do fígado com cirrose comprovada por biópsia, a meia-vida terminal foi de 20,4 horas (intervalo entre 10,8 a 48,0 horas em comparação com 7,4 a 11,0 horas em indivíduos sadios).
Dados de segurança pré-clínicos
A ciclosporina não apresentou evidências de efeitos mutagênicos e teratogênicos em teste padrão de sistemas com aplicação oral (doses orais diárias de até 17 mg/kg em ratos e até 30 mg/kg em coelhos).
Em doses tóxicas (doses orais diárias em ratos de 30 mg/kg e em coelhos de 100 mg/kg), a ciclosporina se mostrou embriotóxica e fetotóxica conforme indicado pelo aumento pré-natal e pós-natal de mortalidade e pela redução do peso fetal, juntamente com relatos de retardo do desenvolvimento esquelético.
Em dois estudos publicados, coelhos expostos a ciclosporina in utero (10 mg/kg/dia subcutâneo) demonstraram redução no número de néfrons, hipertrofia renal, hipertensão sistêmica e progressiva insuficiência renal até 35 semanas de idade.
Ratas grávidas que receberam 12 mg/kg/dia de ciclosporina intravenosa (duas vezes a dose intravenosa humana recomendada) tiveram fetos com incidência aumentada de defeito no septo ventricular.
Estes achados não foram demonstrados em outras espécies e a relevância destes para humanos é desconhecida. Estudos carcinogênicos foram feitos em machos e fêmeas de ratos e camundongos. No estudo de 78 semanas, em camundongos, com doses de 1, 4 e 16 mg/kg/dia, a evidência estatisticamente significativa foi a tendência de linfomas linfocíticos em fêmeas e a incidência de carcinomas hepatocelulares em machos, com a dose intermediária, excedeu significativamente o valor do grupo controle.
No estudo de 24 meses conduzido em ratos, com doses diárias de 0,5, 2 e 8 mg/kg, a incidência de adenomas de células das ilhotas pancreáticas, com dose baixa, excedeu significativamente a do grupo controle. Os carcinomas hepatocelulares e adenomas das ilhotas pancreáticas não apresentaram relação com a dose.
Em estudos com ratos machos e fêmeas, não se observaram efeitos adversos na fertilidade.
A ciclosporina não pareceu ser mutagênica/genotóxica no teste de Ames, no teste V79-HGPRT, nos testes de micronúcleos em camundongos e hamsters chineses, aberrações cromossômicas na medula óssea de hamsters chineses, dominância letal em camundongos e no teste de reparação de DNA em esperma de camundongos tratados.
Um estudo que analisou a troca de cromátides-irmãs (SCE - sister cromatide exchange) induzidas pela ciclosporina, usando-se linfócitos humanos in vitro, indicou efeitos positivos (isto é indução de SCE) com concentrações altas neste sistema.
O aumento da incidência de neoplasia é umas das complicações reconhecidas da imunossupressão em receptores de órgãos transplantados. As formas mais comuns de neoplasmas são os linfomas não-Hodgkin e o carcinoma de pele.
O risco de neoplasia durante o tratamento com ciclosporina é mais alto do que o normal na população saudável, mas similar à dos pacientes que recebem outras terapias imunossupressoras.
Também foi demonstrado que a redução ou descontinuação da terapia imunossupressora pode ocasionar a regressão das lesões.
Cuidados de Armazenamento
Restasis deve ser armazenado em temperatura ambiente (entre 15º e 30º C).
Você deve manter os flaconetes na cama plástica.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Após aberto, deve ser utilizado imediatamente.
Características físicas
Restasis é uma emulsão uniforme branca, opaca ou ligeiramente translúcida.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Todo medicamento deve ser mantido fora do alcance das crianças.