para o que é indicado e para que serve?
Para que serve Câncer de Mama Metastático Perjeta está indicado, em combinação com Herceptin (trastuzumabe) e docetaxel, para pacientes com câncer de mama HER2-positivo metastático ou localmente recorrente não operável, que não tenham recebido tratamento anterior com medicamentos anti-HER2 ou quimioterapia para doença metastática.Continue lendo...
ofertas de
Perjeta 420Mg 14Ml 1 Fras...
ofertas de Perjeta 420Mg 14Ml 1 Fras...
R$ 15.461,99
R$ 15.750,00
R$ 15.790,00
R$ 15.980,00
ATENÇÃO: O texto abaixo deve ser utilizado apenas como uma referência secundária. É um registro histórico da bula, rótulo ou manual do produto. Este texto não pode substituir a leitura das informações que acompanha o produto, cujo fabricante podem mudar a formulação, recomendação, modo de uso e alertas legais sem que sejamos previamente comunicados. Apenas as informações contidas na própria bula, rótulo ou manual que acompanha o produto é que devem estar atualizadas de acordo com a versão comercializada porém, no caso de qualquer dúvida, consulte o serviço de atendimento ao consumidor do produto ou nossa equipe.
Para que serve
Câncer de Mama Metastático
Perjeta está indicado, em combinação com Herceptin (trastuzumabe) e docetaxel, para pacientes com câncer de mama HER2-positivo metastático ou localmente recorrente não operável, que não tenham recebido tratamento anterior com medicamentos anti-HER2 ou quimioterapia para doença metastática.
Tratamento Neoadjuvante de Câncer de Mama
Perjeta está indicado, em combinação com Herceptin (trastuzumabe) e quimioterapia*, para o tratamento neoadjuvante de pacientes com câncer de mama HER2-positivo localmente avançado, inflamatório ou em estágio inicial com elevado risco de recorrência (tanto para > 2 cm de diâmetro quanto para linfonodo positivo) como parte de um esquema terapêutico completo para o câncer de mama inicial.
Como Perjeta funciona?
Perjeta contém um anticorpo monoclonal recombinante humanizado direcionado contra a proteína HER2 da célula de câncer, fazendo com que ela pare de se multiplicar e se autodestrua. Além disso, Perjeta age na toxicidade celular através de determinados anticorpos do organismo.
Perjeta é capaz de inibir sozinho a multiplicação de células tumorais humanas, no entanto, a associação com outros medicamentos, como Herceptin (trastuzumabe) aumenta bastante essa propriedade.
Contraindicação
Perjeta é contraindicado a pacientes com alergia conhecida ao pertuzumabe ou a qualquer outro excipiente da fórmula.
Como usar
Perjeta deve ser utilizado por infusão via intravenosa (ou seja, depois de diluído dentro de uma bolsa de aplicação, deve ser aplicado na veia). O profissional da saúde saberá como preparar o medicamento. A preparação da solução para infusão deverá ser feita por um profissional da saúde, porque é necessário manter técnica asséptica para evitar a contaminação e garantir a esterilidade da solução preparada, uma vez que Perjeta não contém conservantes.
Este medicamento é de uso hospitalar e só poderá ser aplicado por profissionais treinados e habilitados. Seu médico conhece os detalhes da administração e poderá fornecer todas as informações necessárias.
A substituição por qualquer outro medicamento biológico exige o consentimento do médico prescritor.
Posologia
A dose inicial recomendada de Perjeta em combinação com Herceptin (trastuzumabe) e docetaxel
É de 840 mg (2 frascos), infundida durante 60 minutos. Depois disso, deve ser aplicado a cada 3 semanas, em dose de 420 mg (1 frasco), infundida durante um período entre 30 e 60 minutos.
Herceptin (trastuzumabe), que precisa ser usado junto com Perjeta, também deve ser aplicado a cada 3 semanas. A dose inicial é de 8 mg/kg. Depois, a cada 3 semanas, é aplicado na dose de 6 mg/kg.
Os medicamentos devem ser administrados sequencialmente. Perjeta e Herceptin (trastuzumabe) podem ser administrados em qualquer ordem.
Quando estiver recebendo docetaxel, o docetaxel deverá ser administrado depois de Perjeta e Herceptin (trastuzumabe). Um período de observação de 30 a 60 minutos é recomendável após cada infusão de Perjeta e antes do início de qualquer infusão subsequente de trastuzumabe ou docetaxel.
Modificações de dose
Perjeta deve ser descontinuado se o tratamento com Herceptin (trastuzumabe) for descontinuado.
Se o docetaxel for descontinuado, o tratamento com Perjeta e Herceptin (trastuzumabe) pode ser mantido até a progressão da doença ou toxicidade não tratável quando existem metástases.
Reduções de dose não são recomendadas para Perjeta.
Reduções de dose não são recomendadas para Herceptin (trastuzumabe): Vide bula de Herceptin (trastuzumabe) para mais informações.
Para modificações de dose de quimioterápicos é necessário consultar as informações nas bulas dos respectivos produtos.
Duração do tratamento
Instruções especiais de dosagem
O que devo fazer quando eu me esquecer de usar Perjeta?
Se você não puder receber uma das aplicações programadas ou tiver que adiar uma aplicação e o tempo decorrido a partir da data em que a aplicação deveria ser feita for menor que 6 semanas, você deverá receber a aplicação de 420 mg o mais rápido possível.
Se o tempo entre sua última aplicação e a atual for de 6 semanas ou mais, deverá ser reaplicada uma dose de 840 mg em 60 minutos e depois reprogramar as aplicações seguintes com uma dose de 420 mg administrada em um período de 30 a 60 minutos, em intervalos de 3 semanas.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Precauções
Medicamentos que bloqueiam a atividade de HER2, incluindo Perjeta, podem reduzir a fração de ejeção do ventrículo esquerdo, ou seja, podem diminuir a capacidade que o coração tem de bombear sangue para o organismo. Nos estudos clínicos, não foi observada uma redução a ponto de provocar sintomas quando Perjeta foi usado em combinação com Herceptin (trastuzumabe) e docetaxel, em comparação com o uso de placebo associado aos mesmos medicamentos. No entanto, pacientes que receberam radioterapia no tórax ou terapia prévia com antraciclinas tiveram maior risco para desenvolver sintomas cardíacos.
O seu médico precisará solicitar a avaliação da fração de ejeção do ventrículo esquerdo antes do início do tratamento e depois a cada três meses aproximadamente, para verificar se você pode receber Perjeta.
A indicação de Perjeta em combinação com Herceptin (trastuzumabe) e quimioterapia, para o tratamento neoadjuvante de pacientes com câncer, foi baseada em demonstração de uma melhoria na taxa de resposta patológica completa (pCR). Não estão disponíveis dados demonstrando melhora na sobrevida livre de eventos ou sobrevida global.
Perjeta está associado a reações relacionadas à infusão e reações de hipersensibilidade (reações alérgicas) ou anafilaxia (alergia grave que pode levar ao choque e à dificuldade de respiração). Por isso, a aplicação deve ser feita em um local onde você possa ficar em observação entre 30 minutos e uma hora.
Contracepção
Mulheres com possibilidade de engravidar, incluindo aquelas que são parceiras de pacientes do sexo masculino, devem usar métodos contraceptivos efetivos enquanto estiverem recebendo Perjeta e nos 7 meses depois da última dose de Perjeta.
Gravidez e lactação
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Perjeta deve ser evitado durante a gravidez e lactação. Os estudos em animais demonstraram que ele provocou oligoidrâmnio (diminuição do líquido dentro do útero durante a fase de formação dos órgãos) em macacas prenhas, acompanhada de retardo no desenvolvimento dos rins do feto e até óbito do embrião ou feto. Dessa forma, baseado nesses estudos realizados em animais e no mecanismo de ação, é considerado que Perjeta tenha potencial de causar dano ao feto quando administrado em mulheres grávidas. Os anticorpos humanos em geral passam para o leite materno.
Como Perjeta é um anticorpo, existe a possibilidade de que ele passe para o leite materno, e não se sabe quais são os riscos para a criança amamentada com esse leite. Por isso, é preciso optar entre manter o aleitamento ou receber o medicamento. Antes de iniciar o tratamento com
Perjeta, seu médico solicitará exames para verificar ocorrência de gravidez. Se você engravidar durante o uso de Perjeta, um acompanhamento médico cuidadoso deve ser realizado quanto à ocorrência de oligoidrâmnio (pouco líquido amniótico).
Interações medicamentosas
Não foi demonstrada interação entre Perjeta e outros medicamentos usados no tratamento do câncer de mama, como Herceptin (trastuzumabe) e docetaxel. Não há estudos com outros tipos de medicamentos.
Sempre avise a seu médico se estiver tomando qualquer medicamento além dos que foram prescritos por ele, mesmo que sejam à base de ervas (fitoterápicos), homeopáticos ou terapias naturais.
Até o momento, não há informações de que Perjeta possa causar doping. Em caso de dúvidas, consulte o seu médico.
Atenção diabéticos: Contém açúcar.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde.
Reações Adversas
Experiência em estudos clínicos
Uma vez que os estudos clínicos são conduzidos sob condições muito variáveis, a frequências de reações adversas observadas nos ensaios clínicos de um medicamento não podem ser diretamente comparadas com as frequências nos ensaios clínicos de um outro medicamento e podem não refletir as frequências observadas na prática clínica.
Câncer de Mama Mestastático
As reações adversas (RADs) descritas na tabela 1 foram identificadas em 804 pacientes com câncer de mama HER2-positivo tratados no estudo Cleopatra. Pacientes foram randomizados para receber tanto Perjeta em combinação com Herceptin (trastuzumabe) e docetaxel ou placebo em combinação com Herceptin (trastuzumabe) e docetaxel. A duração média do tratamento no estudo foi de 18,1 meses por paciente no grupo tratado com Perjeta e 11,8 meses para os pacientes no grupo tratado com placebo.
Nenhum ajuste de dose foi permitido para Perjeta ou Herceptin (trastuzumabe). As frequências de reações adversas que resultaram em descontinuação definitiva de todos os tratamentos do estudo foram 6,1% para pacientes no grupo tratado com Perjeta e 5,3% para os pacientes no grupo tratado com placebo.
As reações adversas levaram à descontinuação do docetaxel apenas em 23,6% dos pacientes no grupo tratado com Perjeta e 23,2% dos pacientes no grupo tratado com placebo. A Tabela 1 apresenta as reações adversas que ocorreram em pelo menos 10% dos pacientes no grupo tratado com Perjeta. O perfil de segurança de Perjeta permaneceu inalterado com um adicional de 2,75 anos de follow-up (acompanhamento médio total de 50 meses) no estudo Cleopatra.
As reações adversas mais comuns (>30%) observadas com Perjeta em combinação com Herceptin (trastuzumabe) e docetaxel foram diarreia, alopecia, neutropenia, náusea, fadiga, rash e neuropatia periférica. As RADs graus 3-4 do NCI-CTCAE (versão 3.0) mais comuns (> 2%) foram neutropenia, neutropenia febril, leucopenia, diarreia, neuropatia periférica, anemia, astenia e fadiga. Um aumento na incidência de neutropenia febril foi observado em pacientes asiáticos em ambos os braços de tratamento, em comparação com pacientes de outras raças e de outras regiões geográficas. Entre os doentes asiáticos, a incidência de neutropenia febril foi maior no grupo tratado com Perjeta (26%) em comparação com o grupo tratado com placebo (12%).
Tabela 1 – Resumo das RADs muito comuns (> 10%) em pacientes do grupo tratado com Perjeta no estudo Cleopatra
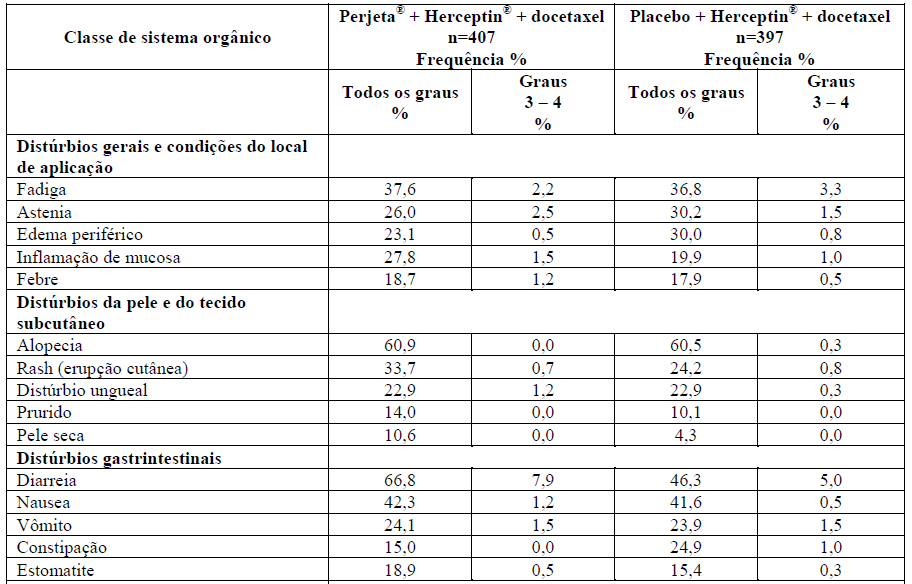
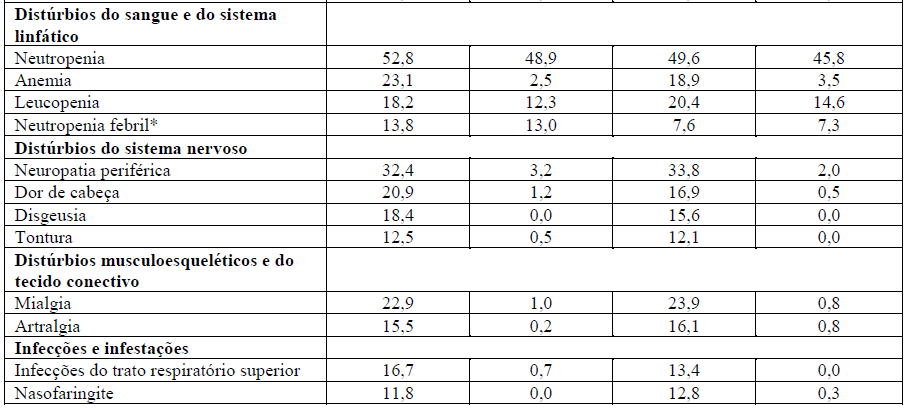
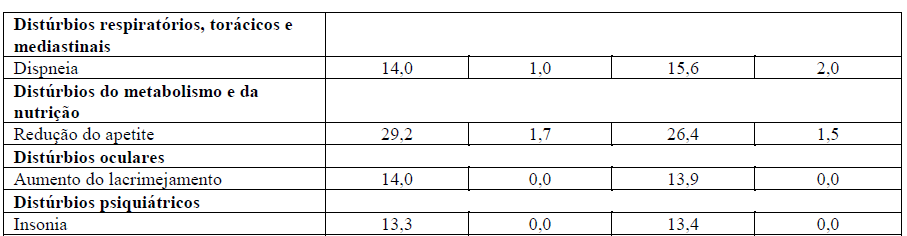
*Nesta tabela, esta indica uma reação adversa que tem sido relatada em associação com um desfecho fatal.
Reações adversas reportadas em pacientes recebendo Perjeta e Herceptin após a descontinuação de docetaxel
No estudo Cleopatra, as reações adversas foram relatadas com menos frequência após a descontinuação do tratamento com docetaxel. Todas as reações adversas no grupo tratado com Perjeta e Herceptin (trastuzumabe) ocorreram em < 10% dos pacientes, com a exceção de diarreia (19,1%), infecção do trato respiratório superior (12,8%), exantema (11,7%), dor de cabeça (11,4%), e fadiga (11,1%).
Tratamento de neoadjuvante de câncer de mama (Neosphere)
No estudo Neosphere, as reações adversas mais comuns observadas com Perjeta em combinação com Herceptin (trastuzumabe) e docetaxel, administrados durante 4 ciclos, foram semelhantes aos observados no grupo tratado com Perjeta no estudo Cleopatra.
As reações adversas mais comuns (> 30%) foram alopecia, neutropenia, diarreia e náusea. As reações adversas NCI-CTCAE (versão 3) graus 3 – 4 mais comuns (> 2%) foram neutropenia, neutropenia febril, leucopenia e diarreia.
Neste grupo, um doente interrompeu o tratamento neoadjuvante permanentemente, devido a um evento adverso. A Tabela 2 relata as reações adversas que ocorreram em pacientes que receberam tratamento neoadjuvante com Perjeta para o câncer de mama no estudo Neosphere.
PerjetaTabela 2 – Resumo das reações adversas muito comuns ? 10% no estudo neoadjuvante em paciente recebendo Perjeta no estudo Neosphere para o câncer de mama no estudo Neosphere
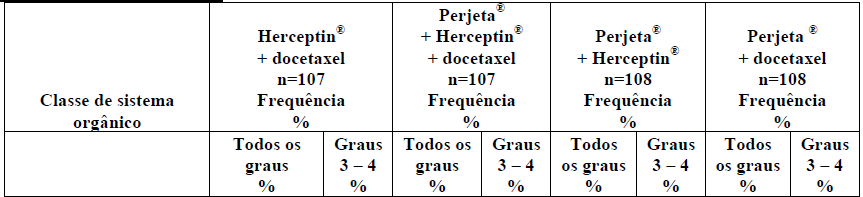
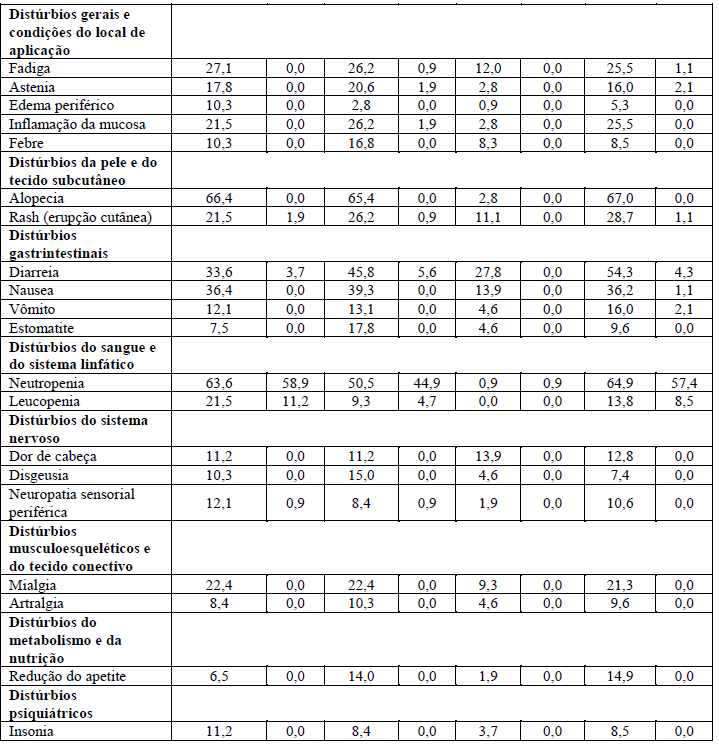
As seguintes reações adversas comuns foram reportadas em < 10% dos pacientes recebendo tratamento neoadjuvante e ocorreram mais frequentemente no grupo de pacientes tratado com Perjeta no estudo Tryphaena: (Ptz= Perjeta; T= Herceptin (trastuzumabe); D=docetaxel; FEC=fluoracil, epirubicina e ciclofosfamida; TCH = docetaxel, carboplatina e Herceptin).
Imunogenicidade
Assim como com todas as proteínas terapêuticas, há um potencial para resposta imunológica ao Perjeta.
Pacientes no estudo pivotal Cforam testados em diversos momentos para a presença de anticorpos antiterapêuticos (ATA) contra Perjeta. Aproximadamente 6,2% (23/372) dos pacientes no braço tratado com placebo e 2,8% (11/386) dos pacientes no braço tratado com Perjeta tiveram teste positivo para ATA. Nenhum dos 34 pacientes, apresentou reações anafiláticas/de hipersensibilidade que fossem claramente relacionadas a ATA. A presença de Perjeta no soro do paciente aos níveis esperados, no momento da amostragem ATA, pode interferir na capacidade do presente ensaio para detectar anticorpos antipertuzumabe. Além disso, o ensaio pode estar detectando anticorpos para o Herceptin.
Como resultado, os dados podem não refletir com precisão a verdadeira incidência de desenvolvimento de anticorpos antipertuzumabe.
Resultados de ensaio de imunogenicidade são altamente dependentes de diversos fatores, incluindo ensaio de sensibilidade e especificidade, metodologia de ensaio, manipulação de amostras, momento da coleta de amostra, medicamentos concomitantes e doença subjacente. Por essas razões, a comparação de incidência de anticorpos contra Perjeta com a incidência de anticorpos contra outros produtos pode não ser clara.
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, informe seu médico ou cirurgião-dentista.
Composição
Cada frasco-ampola de uso único com 14 mL contém:
420 mg de pertuzumabe.
Excipientes: Ácido acético, histidina, polissorbato 20, sacarose e água para injetáveis.
Superdosagem
Não há nenhuma experiência com superdose em estudos clínicos humanos. Doses únicas acima de 25 mg/kg (1.727 mg) não foram testadas.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível. Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação Medicamentosa
Um subestudo que incluiu 37 pacientes do estudo pivotal CLEOPATRA demonstrou que não há evidência de interação medicamentosa entre Pertuzumabe (substância ativa deste medicamento) e Herceptin (trastuzumabe) e entre Pertuzumabe (substância ativa deste medicamento) e docetaxel.
Além disso, nenhuma interação farmacocinética clínica relevante de docetaxel ou Herceptin (trastuzumabe) coadministrados com Pertuzumabe (substância ativa deste medicamento) foi evidenciada com base na análise de farmacocinética populacional.
Os dados farmacocinéticos do estudo NEOSPHERE confirmam não haver interação medicamentosa entre Pertuzumabe (substância ativa deste medicamento) e os demais agentes. Esta ausência de interação medicamentosa foi confirmada por dados farmacocinéticos obtidos no estudo NEOSPHERE.
Quatro estudos avaliaram os efeitos de Pertuzumabe (substância ativa deste medicamento) sobre a farmacocinética de agentes citotóxicos coadministrados, docetaxel, gencitabina, erlotinibe e capecitabina. Não houve evidência de nenhuma interação farmacocinética entre Pertuzumabe (substância ativa deste medicamento) e quaisquer desses agentes.
A farmacocinética de Pertuzumabe (substância ativa deste medicamento) nesses estudos foi comparável à observada em estudos com monoterapia.
Ação da Substância
Resultados da Eficácia
Câncer de mama metastático
Pertuzumabe (substância ativa deste medicamento) em combinação com Herceptin (trastuzumabe) e docetaxel.
CLEOPATRA é um estudo clínico Fase III, multicêntrico, randomizado, duplo-cego e controlado com placebo que incluiu 808 pacientes com câncer de mama HER2-positivo metastático ou localmente recorrente não ressecável, que não receberam tratamento prévio com medicamentos anti-HER2 ou quimioterapia para doença metastática.
Foram solicitadas amostras de tumor mamário para demonstrar a superexpressão de HER2, definida como um escore de 3+ por razão de amplificação IHQ (imunohistoquímica) ou ISH (hibridização in situ) ? 2,0, conforme determinação de um laboratório central. Os pacientes foram randomizados 1:1 para receber placebo mais Herceptin (trastuzumabe) e docetaxel ou Pertuzumabe (substância ativa deste medicamento) mais Herceptin (trastuzumabe) e docetaxel.
A randomização foi estratificada por tratamento prévio (terapia de novo ou terapia adjuvante/neoadjuvante prévia) e região geográfica (Europa, América do Norte, América do Sul e Ásia). Pacientes com terapia adjuvante ou neoadjuvante prévia precisavam ter um intervalo livre de doença de pelo menos 12 meses antes da inclusão no estudo.
Pertuzumabe (substância ativa deste medicamento) foi administrado por via intravenosa (IV) em dose inicial de 840 mg, seguida por uma dose de 420 mg. A administração foi feita a cada três semanas. Herceptin (trastuzumabe) foi administrado intravenosamente em dose inicial de 8 mg/kg, seguida por 6 mg/kg, igualmente a cada três semanas. Os pacientes foram tratados com Pertuzumabe (substância ativa deste medicamento) e Herceptin (trastuzumabe) até a progressão da doença, retirada de consentimento ou toxicidade não manejável. O docetaxel foi administrado em uma dose inicial de 75 mg/m2 por infusão IV a cada 3 semanas, durante, pelo menos, 6 ciclos. A dose de docetaxel poderia ser escalonada até 100 mg/m2 a critério do investigador se a dose inicial fosse bem tolerada.
No momento da análise primária, o número médio de ciclos de tratamento em estudo recebidos foi de 16,2 no braço de tratamento com placebo e 19,9 no braço tratado com Pertuzumabe (substância ativa deste medicamento).
O desfecho primário do estudo foi a sobrevida livre de progressão (SLP) de acordo com avaliação de um comitê de revisão independente (CRI) e definido como o tempo desde a data de randomização até a data da progressão de doença ou óbito (por qualquer causa), se ocorrer óbito dentro de 18 semanas da última avaliação tumoral. Desfechos secundários de eficácia foram sobrevida global (SG), avaliação do investigador (SLP), taxa de resposta objetiva (TRO), duração de resposta e tempo até a progressão dos sintomas de acordo com o questionário FACT B QoL.
Dados demográficos foram bem equilibrados [idade mediana de 54 anos, maioria branca (59%) e todos os pacientes do sexo feminino, com exceção de 2]. Aproximadamente metade dos pacientes em cada braço de tratamento apresentava doença positiva para receptor hormonal [definida como receptor de estrógeno (RE) positivo e/ou receptor de progesterona (RPg) positivo] e aproximadamente metade dos pacientes em cada braço de tratamento tinha recebido terapia adjuvante ou neoadjuvante prévia [192 pacientes (47,3%) no braço tratado com placebo vs. 184 pacientes (45,8%) no braço tratado com Pertuzumabe (substância ativa deste medicamento)].
No momento da análise primária da sobrevida livre de progressão, um total de 242 pacientes (59%) no braço tratado com placebo e 191 pacientes (47,5%) no braço tratado com Pertuzumabe (substância ativa deste medicamento) apresentava doença progressiva confirmada pelo CRI ou morreu dentro de 18 semanas a partir da última avaliação tumoral.
No momento da análise primária, o estudo CLEOPATRA demonstrou aumento estatisticamente significativo da SLP avaliada pelo CRI [razão de risco (HR) = 0,62, IC 95% = 0,51, 0,75, p < 0,0001] no braço tratado com Pertuzumabe (substância ativa deste medicamento) em comparação com o braço tratado com placebo, e um aumento de SLP mediana de 6,1 meses (SLP mediana de 12,4 meses no braço tratado com placebo vs. 18,5 meses no braço tratado com Pertuzumabe (substância ativa deste medicamento)) (vide Figura 1). Os resultados para SLP avaliada pelo investigador foram comparáveis aos observados para SLP avaliada pelo CRI (SLP mediana foi de 12,4 meses para placebo vs. 18,5 meses para Pertuzumabe (substância ativa deste medicamento)) (vide Tabela 1). Resultados compatíveis foram observados entre os subgrupos de pacientes predeterminados, incluindo os subgrupos baseados em fatores de estratificação por região geográfica e terapia adjuvante/neoadjuvante prévia ou câncer de mama metastático de novo (vide Figura 2).
Os resultados de eficácia do estudo CLEOPATRA estão resumidos na Tabela 1:
Tabela 1 - Resumo da eficácia do estudo CLEOPATRA
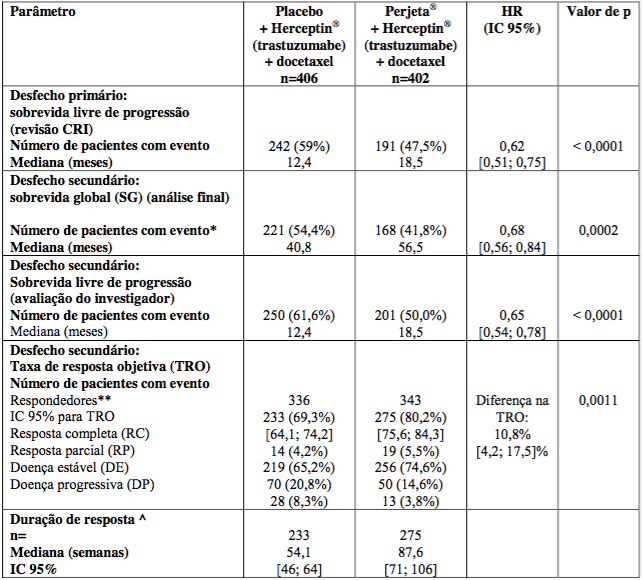
* Análise final de sobrevida global, data de corte 11 de fevereiro de 2014.
** Pacientes com melhor resposta global entre RC ou RP confirmadas por RECIST.
^ Avaliada em pacientes com melhor resposta global entre RC ou RP.
Taxa de resposta objetiva e duração de resposta são baseadas em avaliações tumorais avaliadas por CRI.
Figura 1: Curva de Kaplan-Meier de sobrevida livre de progressão avaliada pelo CRI
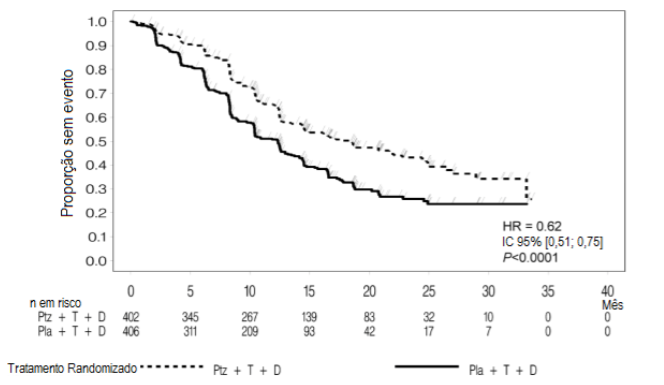
D = docetaxel; HR = hazard ratio; Ptz = pertuzumabe (Pertuzumabe (substância ativa deste medicamento)); T = trastuzumabe (Herceptin); Pla = placebo
Figura 2: SLP avaliada pelo CRI por subgrupo de pacientes
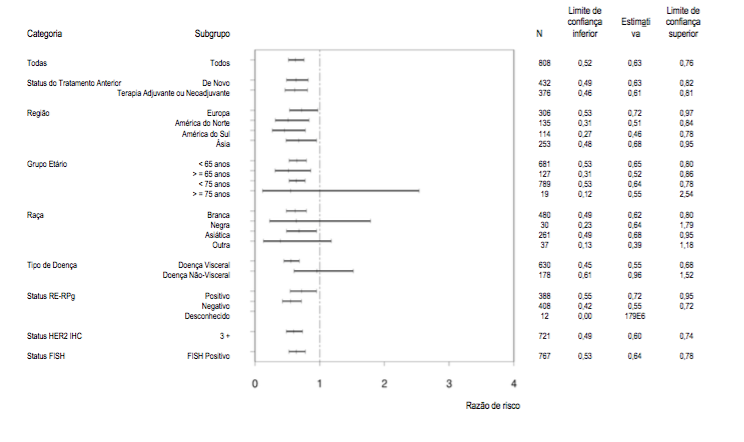
Na análise primária de eficácia, uma análise interina de SG demonstrou uma forte tendência sugestiva de um benefício de sobrevida favorável ao braço tratado com Pertuzumabe (substância ativa deste medicamento).
Uma análise interina de SG realizada um ano depois da análise primária de eficácia demonstrou um benefício de SG estatisticamente significativo favorável ao braço tratado com Pertuzumabe (substância ativa deste medicamento) (HR 0,66, p = 0,0008 em teste de log-rank). O tempo mediano até óbito foi de 37,6 meses no braço tratado com placebo, mas ainda não tinha sido atingido no braço tratado com Pertuzumabe (substância ativa deste medicamento).
A análise final da SG foi realizada quando os 389 pacientes foram a óbito (221 no grupo tratado com placebo e 168 no grupo tratado com Pertuzumabe (substância ativa deste medicamento)). O benefício de SG estatisticamente significativo a favor do grupo tratado com Pertuzumabe (substância ativa deste medicamento) foi mantido (HR 0,68, p = 0,0002 em teste de log-rank). O tempo mediano até óbito foi de 40,8 meses no grupo tratado com placebo e 56,5 meses no grupo tratado com Pertuzumabe (substância ativa deste medicamento) (vide Tabela 2 e Figura 3).
Figura 3: Curva de Kaplan-Meier para sobrevida global
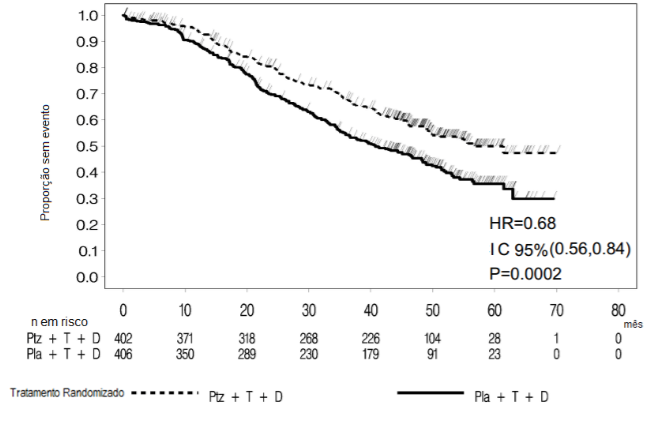
D = docetaxel; HR = hazard ratio; Ptz = pertuzumabe (Pertuzumabe (substância ativa deste medicamento)); T = trastuzumabe (Herceptin); Pla = placebo.
Não houve diferença estatisticamente significativa entre os braços de tratamento quando avaliada Qualidade de Vida Relacionada à Saúde, conforme avaliação pelo tempo até progressão de sintomas sobre a subescala FACT-B TOI-PFB, definida como uma redução de 5 pontos no escore de subescala (HR = 0,97, IC 95% 0,81; 1,16).
Em uma análise exploratória, pacientes tratados com Pertuzumabe (substância ativa deste medicamento) em combinação com Herceptin (trastuzumabe) e docetaxel apresentaram menor risco de progressão sintomática em subescala FACT-B de câncer de mama (definido como uma redução de 2 pontos no escore da subescala) em comparação com os tratados com Herceptin (trastuzumabe) e docetaxel apenas (HR = 0,78, IC 95% = 0,65; 0,94).
BO17929
BO17929 foi um estudo não randomizado Fase II de braço único com Pertuzumabe (substância ativa deste medicamento) e foi conduzido incluindo pacientes com câncer de mama metastático (CMM) HER2-positivo que tinham recebido tratamento prévio com Herceptin (trastuzumabe). O estudo foi dividido em 3 coortes.
Coortes 1 e 2
66 pacientes nas coortes 1 e 2 receberam pelo menos uma dose de Pertuzumabe (substância ativa deste medicamento) e Herceptin (trastuzumabe) (toda população tratada e todos os pacientes tinham recebido tratamento prévio para doença metastática; metade estava recebendo tratamento de segunda linha para doença metastática, enquanto que 35% estavam recebendo tratamento de terceira linha ou posterior. Além disso, 71% tinham recebido quimioterapia neoadjuvante). No momento da análise primária, a duração mediana de tratamento em estudo foi de nove ciclos (27 semanas). No momento da análise primária, TRO e TBC (taxa de benefício clínico) são apresentadas na Tabela 2. A SLP mediana e tempo para progressão (TPP) foram de 24 semanas. O tempo mediano até a resposta foi de 11 semanas e, naqueles pacientes com resposta, a duração mediana de resposta foi de 25 semanas.
Coorte 3:
29 pacientes receberam pelo menos um ciclo de Pertuzumabe (substância ativa deste medicamento). Desses 29 pacientes, 12 participaram apenas da Fase de agente único e 17 prosseguiram para receber Pertuzumabe (substância ativa deste medicamento) e Herceptin (trastuzumabe) quando apresentaram progressão documentada recebendo Pertuzumabe (substância ativa deste medicamento) apenas.
Todos os 29 pacientes progrediram durante terapia de primeira linha no braço metastático e 41,4% também progrediram depois da terapia de segunda linha. Todos os pacientes na Coorte 3 receberam pelo menos uma dose completa de Pertuzumabe (substância ativa deste medicamento). Pacientes em tratamento com Pertuzumabe (substância ativa deste medicamento) e Herceptin (trastuzumabe) receberam uma mediana de 12 ciclos no total.
A Tabela 2 mostra que Pertuzumabe (substância ativa deste medicamento) sozinho apresentou atividade modesta em pacientes depois de falência de Herceptin (trastuzumabe) (coluna do meio). Essas respostas ocorreram em pacientes cuja doença progrediu recentemente com cada anticorpo quando administrado separadamente. Além disso, os 3 pacientes tinham doença estável com seis meses ou mais para uma taxa de benefício clínico total de 35,3%.
Tabela 2 - Estudo BO17929: Dados de eficácia
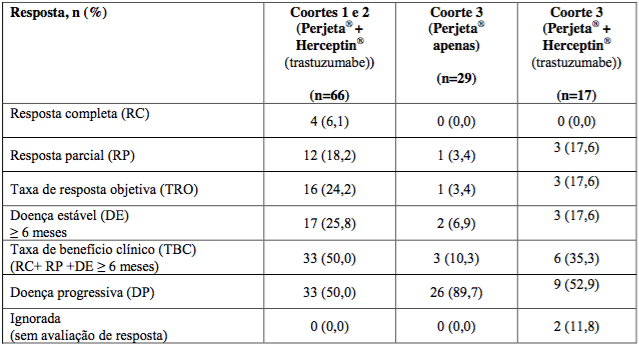
Nota: > 6 meses = 8 ciclos de terapia.
Tratamento Neoadjuvante de Câncer de Mama
A indicação é baseada em dois estudos, NEOSPHERE e TRYPHAENA. Esses dois estudos clínicos Fase II, multicêntricos, randomizados, incluiram pacientes com câncer de mama HER2-positivo ressecável, localmente avançado ou inflamatório (T2-4d) elegíveis para terapia neoadjuvante.
Em pacientes com tratamento neoadjuvante, tumores de mama localmente avançados e inflamatórios são considerados de alto risco, independente do status dos receptores hormonais. No estágio inicial do câncer de mama, o tamanho do tumor, grau, status dos receptores de hormônio e mestástases linfonodais devem ser levados em conta na hora da avaliação de risco.
NEOSPHERE (WO20697)
NEOSPHERE é um estudo clínico Fase II, multicêntrico, randomizado, conduzido com 417 pacientes com câncer de mama HER2-positivo recém diagnosticadas, localmente avançado, ressecável, ou inflamatório (T2-4d; tumor primário > 2 cm em diâmetro), elegíveis para terapia neoadjuvante. Pacientes com mestástases, câncer de mama bilateral, importantes fatores de risco cardíacos ou LVEF < 55% não foram incluídos.
A maioria das pacientes tinha menos de 65 anos de idade. Foram exigidas amostras tumorais para garantir superexpressão de HER2, definida como escore de 3+ por razão de IHQ (imunohistoquímica) ou amplificação através da ISH (hibridização in situ) ? 2,0 por laboratório central.
Os pacientes foram randomizados para receber um de quatro esquemas neoadjuvantes antes da cirurgia, como se segue: Herceptin (trastuzumabe) mais docetaxel, Pertuzumabe (substância ativa deste medicamento) mais Herceptin (trastuzumabe) e docetaxel, Pertuzumabe (substância ativa deste medicamento) mais Herceptin (trastuzumabe) ou Pertuzumabe (substância ativa deste medicamento) mais docetaxel. A randomização foi estratificada por tipo de câncer de mama (ressecável, localmente avançado ou inflamatório) e positividade para receptor de estrógeno (RE) ou de progesterona (RP).
Pertuzumabe (substância ativa deste medicamento) foi administrado por via intravenosa em dose inicial de 840 mg, seguida por 420 mg a cada três semanas durante quatro ciclos. Trastuzumabe foi administrado por via intravenosa em dose inicial de 8 mg/kg, seguida por 6 mg/kg a cada três semanas durante quatro ciclos. O docetaxel foi administrado como uma dose inicial de 75 mg/m2 por infusão intravenosa, a cada 3 semanas por 4 ciclos.
A dose de docetaxel pôde ser escalonada até 100 mg/m2, a critério do investigador, se a dose foi bem tolerada. Depois da cirurgia, todos os pacientes receberam três ciclos de 5- fluorouracil (600 mg/m2), epirrubicina (90 mg/m2), ciclofosfamida (600 mg/m2) (FEC) administrados via intravenosa a cada três semanas e Herceptin (trastuzumabe) administrado via intravenosa a cada três semanas até completar um ano de terapia.
Os pacientes no braço Pertuzumabe (substância ativa deste medicamento) mais Herceptin (trastuzumabe) e docetaxel receberam docetaxel a cada três semanas durante quatro ciclos antes do FEC depois da cirurgia, de forma que todos os pacientes receberam doses cumulativas equivalentes de agentes quimioterápicos e Herceptin (trastuzumabe).
O desfecho primário do estudo foi a resposta patológica completa (rPC) na mama (ypT0/is). Os desfechos secundários de eficácia foram porcentagem de resposta clínica, porcentagem de cirurgia conservadora de mama (T2-3 apenas), sobrevida livre de doença (SLD) e sobrevida livre de progressão (SLP). Porcentagens de pCR exploratório adicionais incluíram estado linfonodal (ypT0/isN0 e ypT0N0).
Os dados demográficos foram bem equilibrados (a mediana da idade foi de 49 a 50 anos, a maioria era branca (71%) e todos os pacientes eram do sexo feminino). No total, 7% das pacientes apresentava câncer de mama inflamatório, 32%, câncer de mama localmente avançado e 61%, câncer de mama ressecável. Aproximadamente metade das pacientes em cada grupo de tratamento apresentava doença positiva para receptor de hormônio (definida como RE positivo e/ou RP positivo).
Os resultados de eficácia estão resumidos na Tabela 3. Observou-se ganho estatisticamente significativo e clinicamente relevante no percentual de rPC para pacientes recebendo Pertuzumabe (substância ativa deste medicamento) mais Herceptin (trastuzumabe) e docetaxel em comparação com pacientes recebendo Herceptin (trastuzumabe) e docetaxel (45,8% vs. 29,0%, valor de p = 0,0141). Foi observado um padrão constante de resultados, independentemente da definição de rPC.
Percentuais de resposta patológica completa (rPC), bem como magnitude do ganho com Pertuzumabe (substância ativa deste medicamento), foram menores no subgrupo de pacientes com tumores positivos para receptor hormonal do que em pacientes com tumores negativos para receptor hormonal: 12,0% [IC 95% (4,5%, 24,3%)], 22,0% [IC 95% (11,5%, 36,0%)], 2,0 %[IC 95% (0,1%, 10,5%)], 8,7% [IC 95% (2,4%, 20,8%)], nos tumores positivos para receptor hormonal, e 29,8% [IC 95% (18,4%, 43,4%)], 54,4% [IC 95% (40,7%, 67,6%)], 20,0% [IC 95% (10,4%, 33,0%)], 26,0% [IC 95% (14,6%, 40,3%)], nos tumores negativos para receptor hormonal.
TRYPHAENA (BO22280)
TRYPHAENA é um estudo clínico Fase II, multicêntrico, randomizado, conduzido com 225 pacientes com câncer de mama HER2-positivo localmente avançado, ressecável ou inflamatório (T2-4d; tumor primário > 2 cm em diâmetro), que não receberam tratamento prévio com Herceptin (trastuzumabe), quimioterapia ou radioterapia. Pacientes com mestástases, câncer de mama bilateral, importantes fatores de risco cardíacos ou LVEF < 55% não foram incluídos. Amostras de tumor de mama deveriam apresentar superexpressão de HER2, definida como um escore de 3+ através da IHQ (imunohistoquímica) ou amplificação através de ISH (hibridização in situ) ? 2,0 por laboratório central.
Os pacientes foram randomizados para receber um de três esquemas neoadjuvantes antes da cirurgia, como se segue: três ciclos de FEC seguidos por três ciclos de docetaxel, todos em combinação com Pertuzumabe (substância ativa deste medicamento) e Herceptin (trastuzumabe), três ciclos de FEC isoladamente seguidos por três ciclos de docetaxel e Herceptin (trastuzumabe) em combinação com Pertuzumabe (substância ativa deste medicamento) ou seis ciclos de TCH em combinação com Pertuzumabe (substância ativa deste medicamento). A randomização foi estratificada por câncer de mama tipo (ressecável, localmente avançado ou inflamatório) e positividade para RE e/ou RP.
Pertuzumabe (substância ativa deste medicamento) foi administrado por via intravenosa em dose inicial de 840 mg, seguida por 420 mg a cada três semanas. Herceptin (trastuzumabe) foi administrado via intravenosa em dose inicial de 8 mg/kg, seguida por 6 mg/kg a cada três semanas. 5-Fluoruracil (500 mg/m2), epirrubicina (100 mg/m2), ciclofosfamida (600 mg/m2) foram administrados por via intravenosa a cada três semanas durante três ciclos.
Docetaxel foi administrado em dose inicial de 75 mg/m2 em infusão IV a cada três semanas, com a opção de escalonamento para 100 mg/m2 a critério do investigador se a dose inicial for bem tolerada. No entanto, no braço Pertuzumabe (substância ativa deste medicamento) em combinação com TCH, docetaxel foi administrado por via intravenosa na dose de 75 mg/m2, nenhum escalonamento foi permitido e carboplatina (AUC 6) foi administrada a cada três semanas. Depois da cirurgia, todos os pacientes receberam Herceptin (trastuzumabe) para completar um ano de terapia, que foi administrado via intravenosa a cada três semanas.
O desfecho primário deste estudo foi segurança cardíaca durante o período de tratamento neoadjuvante do estudo.
Desfechos secundários de eficácia foram taxa de rPC na mama (ypT0/is), SLD, SLP e SG.
As características demográficas foram bem equilibradas (a mediana da idade foi de 49 a 50 anos, a maioria era branca (77%) e todas eram do sexo feminino). No total, 6% das pacientes apresentavam câncer de mama inflamatório, 25% apresentavam câncer de mama localmente avançado e 69% apresentavam câncer de mama ressecável; aproximadamente metade das pacientes em cada grupo de tratamento apresentava doença RE-positivo e/ou RP-positivo.
Taxas elevadas de rPC foram observadas nos três braços de tratamento (veja Tabela 3). Observou-se um padrão constante de resultados independentemente da definição de rPC. As taxas de rPC foram mais baixas no subgrupo de pacientes com tumores positivos para receptor hormonal do que em pacientes com tumores negativos para este receptor 41,0% [IC 95% (25,6%, 57,9%)], 45,7% [IC 95% (28,8%, 63,4%)], e 47,5% [IC 95% (31,5%, 63,9%)], para tumores positivos para receptor hormonal, e 73,5% [IC 95% (55,6%, 87,1%)], 62,5% [IC 95% (45,8%, 77,3%)] e 81,1% [IC 95% (64,8%, 92,0%)], para tumores negativos para receptor hormonal.
Tabela 3: NEOSPHERE (WO20697) e TRYPHAENA (BO22280): Resumo de eficácia (população ITT)
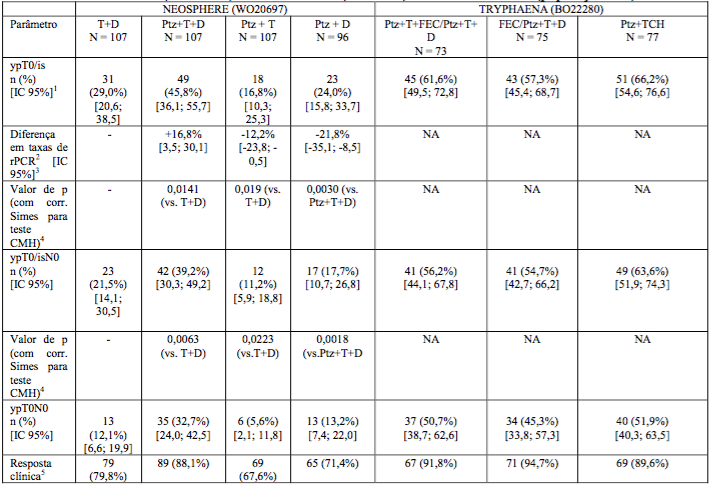
Chaves de abreviações (Tabela 3):
T: Herceptin (trastuzumabe); D: docetaxel; Ptz: Pertuzumabe (substância ativa deste medicamento); FEC: 5-fluorouracil, epirrubicina, ciclofosfamida, TCH: docetaxel, carboplatina e trastuzumabe.
- IC 95% para uma amostra binomial us+.ando método de Pearson-Clopper.
- O tratamento Ptz+T+D e Ptz+T são comparados com T+D enquanto que Ptz+D é comparado com Ptz+T+D.
- IC 95% aproximado para diferença de duas taxas usando método Hauck-Anderson.
- Valor de p pelo teste de Cochran-Mantel-Haenszel com ajuste para multiplicidade Simes.
- ?5. Resposta clínica representa pacientes com melhor resposta global incluindo RC ou RP durante o período neoadjuvante (na lesão de mama primária).
Características Farmacológicas
Farmacodinâmica
Mecanismo de ação
Pertuzumabe (substância ativa deste medicamento) é um anticorpo monoclonal recombinante humanizado que age seletivamente sobre o domínio extracelular de dimerização (subdomínio II) do receptor-2 do fator de crescimento epidérmico humano (HER2). Dessa forma, ele bloqueia a heterodimerização ligante dependente do HER2 com outros membros da família HER, incluindo EGFR, HER3 e HER4.
Como resultado, Pertuzumabe (substância ativa deste medicamento) inibe a sinalização intracelular iniciada por ligante, por meio de duas vias de sinais importantes, a de proteína quinase ativada por mitógeno (MAP) e fosfoinositide-3quinase (PI3K). A inibição dessas vias de sinalização pode resultar em parada de crescimento celular e apoptose, respectivamente. Além disso, Pertuzumabe (substância ativa deste medicamento) é o mediador de citotoxicidade celular dependente de anticorpos (CCDA).
Embora Pertuzumabe (substância ativa deste medicamento) isoladamente iniba a proliferação de células tumorais humanas, a combinação de Pertuzumabe (substância ativa deste medicamento) e Herceptin (trastuzumabe) aumentou significativamente a atividade antitumoral em modelos de xenoenxerto com superexpressão de HER2.
Farmacocinética
Em vários estudos clínicos em diversas indicações, não houve alteração no clearance (CL) de pertuzumabe em doses de 2-25 mg/kg. Com base em uma análise farmacocinética (PK) populacional que incluiu 481 pacientes, o clearance mediano de pertuzumabe foi de 0,235 L/dia e a meia-vida mediana foi de 18 dias.
A análise da PK populacional sugeriu que não há diferenças de PK com base na idade, sexo e etnia (japonesa vs. não japonesa). A albumina e peso corpóreo basais foram as covariáveis mais significativas que influenciaram o CL. O clearance diminuiu em pacientes com maiores concentrações basais de albumina e aumentou em pacientes com maior peso corpóreo.
No entanto, análises de sensibilidade realizadas na dose e esquemas recomendados de Pertuzumabe (substância ativa deste medicamento) mostraram que nos valores extremos dessas duas covariáveis não houve nenhum impacto significativo sobre a capacidade de atingir concentrações-alvo em equilíbrio dinâmico identificadas nos modelos pré-clínicos de xenoenxerto tumoral. Portanto, não há necessidade de ajustar a dose de pertuzumabe com base nessas covariáveis.
Os resultados farmacocinéticos do estudo NEOSPHERE foram compatíveis com as previsões do modelo populacional anterior.
Farmacocinética em situações clínicas especiais
Dados de segurança pré-clínica
Mutagenicidade
Não foram realizados estudos para avaliar o potencial mutagênico de pertuzumabe.
Cuidados de Armazenamento
Perjeta em frasco-ampola deve ser conservado sob refrigeração (entre 2 e 8ºC) e dentro de sua embalagem original para proteger da luz. Não congele e não agite.
O profissional da saúde saberá como armazenar o medicamento depois de aberto.
Número de lote e datas de fabricação e validade: vide embalagem. Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Após preparo, a solução diluída deve ser utilizada imediatamente.
Característíca física
A solução de Perjeta apresenta coloração incolor a castanho claro e é clara a levemente opalescente.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Todo medicamento deve ser mantido fora do alcance das crianças.
Dizeres Legais
MS-1.0100.0657
Farm. Resp.:
Tatiana Tsiomis Díaz - CRF-RJ nº 6942
Fabricado para
F. Hoffmann-La Roche Ltd, Basileia, Suíça
Por Roche Diagnostics GmbH, Mannheim, Alemanha
Embalado por
F. Hoffmann-La Roche Ltd, Kaiseraugst, Suíça
Registrado, importado e distribuído no Brasil por
Produtos Roche Químicos e Farmacêuticos S.A.
Est. dos Bandeirantes, 2020 CEP 22775-109 – Rio de Janeiro – RJ
CNPJ 33.009.945/0023-39
Serviço Gratuito de Informações - 0800 7720 289
www.roche.com.br
Venda sob prescrição médica.
Uso restrito a hospitais.
informações complementares
| Fabricante |
| ROCHE |
| Princípio ativo |
| Pertuzumabe |
| Categoria do medicamento |
| Medicamentos Especiais |