Comparamos o preço de Sucrofer - 20 Mg/Ml Solução Injetável 5 Ampola 5 Ml, veja o menor preço

RReferência
Para que serve
PARA QUE ESTE MEDICAMENTO É INDICADO SUCROFER é indicado para reposição de ferro, nos casos em que não é possível a administração oral por problemas na absorção ou intolerâncias a preparações orais de ferro 2
Contraindicação
QUANDO NÃO DEVO USAR ESTE MEDICAMENTO SUCROFER é contraindicado para pacientes com evidência de sobrecarga de ferro, com hipersensibilidade conhecida ao ferro, complexos de ferro ou qualquer excipiente e com anemia não causada por deficiência de ferro A segurança e eficácia de SUCROFER não foram estabelecidas em pacientes pediátricos Este medicamento é contraindicado para uso por crianças 4
Como usar
recomendadas para administração (ver COMO DEVO USAR ESTE MEDICAMENTO ) Efeito sobre a capacidade de dirigir veículos e operar máquinas 2 É improvável que SUCROFER tenha alguma influência na capacidade de dirigir ou operar máquinas Uso durante a gravidez e amamentação Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica Não se sabe se SUCROFER passa para o leite materno, porém, uma vez que muitos fármacos aparecem no leite humano recomenda-se cautela quando SUCROFER for administrado e você estiver amamentando SUCROFER não deve ser administrado concomitantemente a preparações orais de ferro, pois a absorção oral é reduzida Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento Não use medicamento sem o conhecimento do seu médico Pode ser perigoso para a sua saúde Os estudos realizados com SUCROFER, de forma geral, não foram observadas diferenças em relação à segurança entre idosos e os pacientes mais jovens, porém, a maior sensibilidade de sujeitos mais velhos não pode ser desconsiderada 5
COMO DEVO USAR ESTE MEDICAMENTO Modo de Usar SUCROFER deve ser administrado exclusivamente em hospitais, por via intravenosa (injeção lenta ou infusão) SUCROFER deve ser administrado de acordo com as recomendações descritas na bula do profissional da saúde As suas doses de SUCROFER serão determinadas pelo médico, levando em consideração a sua necessidade de ferro, seu peso e a sua reserva de ferro Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento Não interrompa o tratamento sem o conhecimento do seu médico 3 7
O QUE DEVO FAZER QUANDO EU ME ESQUECER DE USAR ESTE MEDICAMENTO Não aplicável, pois SUCROFER deve ser administrado exclusivamente em hospitais Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista 8
Precauções
O QUE DEVO SABER ANTES DE USAR ESTE MEDICAMENTO Seu médico deverá acompanhar parâmetros do sangue e da hemoglobina enquanto você estiver usando SUCROFER Se houver sinal de sobrecarga de ferro, o tratamento com SUCROFER deve ser suspenso SUCROFER pode causar reações alérgicas leves a moderadas Hipotensão (queda de pressão) foi relatada em pacientes fazendo hemodiálise (tratamento para remoção de substâncias do sangue) e recebendo ferro intravenoso Seu médico seguir com cautela as instruções
Reações Adversas
QUAIS OS MALES QUE ESTE MEDICAMENTO PODE ME CAUSAR As reações adversas mais frequentemente reportadas nos estudos clínicos realizados com SUCROFER foram: mudança temporária do paladar, hipotensão (queda de pressão), febre e calafrios, reações no local da injeção e náusea, ocorrendo em 0,5% a 1,5% dos pacientes Reações anafilactoides (semelhantes à alergia) não graves ocorreram raramente De modo geral, reações anafilactoides são, potencialmente, as reações adversas mais graves que podem ocorrer Nos estudos clínicos, as seguintes reações adversas medicamentosas foram reportadas com relação temporal à administração de SUCROFER, tendo pelo menos uma relação causal possível Distúrbios do sistema nervoso central Reações comuns (ocorrem entre 1% e 10% dos pacientes que utilizam este medicamento): alteração temporária do paladar (em particular, gosto metálico) Reações incomuns (ocorrem entre 0,1% e 1% dos pacientes que utilizam este medicamento): dor de cabeça, tontura Reações raras (ocorrem entre 0,01% e 0,1% dos pacientes que utilizam este medicamento): parestesia (sensação de formigamento, picada ou queimadura não causada por estímulos externos), síncope (perda abrupta e temporária de consciência e da postura), perda de consciência, sensação de queimação Distúrbios cardiovasculares Reações incomuns (ocorrem entre 0,1% e 1% dos pacientes que utilizam este medicamento): hipotensão e colapso, taquicardia (aumento da frequência cardíaca) e palpitações Reações raras (ocorrem entre 0,01% e 0,1% dos pacientes que utilizam este medicamento): hipertensão (aumento de pressão) Distúrbios respiratórios, torácicos e mediastinais Reações incomuns (ocorrem entre 0,1% e 1% dos pacientes que utilizam este medicamento): broncoespasmo (estreitamento dos brônquios, causando dificuldade de respiração), dispneia (falta de ar) Distúrbios gastrointestinais Reações incomuns (ocorrem entre 0,1% e 1% dos pacientes que utilizam este medicamento): náusea, vômitos, dor abdominal e diarreia Distúrbios da pele e tecido subcutâneo Reações incomuns (ocorrem entre 0,1% e 1% dos pacientes que utilizam este medicamento): prurido, urticária, rash, exantema (erupção cutânea) e eritema (lesões avermelhadas e salientes na pele) Distúrbios musculoesqueléticos, do tecido conectivo e ósseos Reações incomuns (ocorrem entre 0,1% e 1% dos pacientes que utilizam este medicamento): câimbras musculares, mialgia (dores musculares) 4 Distúrbios gerais e no local da administração Reações incomuns (ocorrem entre 0,1% e 1% dos pacientes que utilizam este medicamento): febre, calafrios, rubor, dor e aperto no peito, flebite superficial (inflamação do vaso sanguíneo), queimação e inchaço Reações raras (ocorrem entre 0,01% e 0,1% dos pacientes que utilizam este medicamento): artralgia (dor nas articulações), edema periférico (inchaço nas extremidades do corpo), fadiga (cansaço), astenia (fraqueza), mal estar, sensação de calor, edema (inchaço) Distúrbios do sistema imune Reações raras (ocorrem entre 0,01% e 0,1% dos pacientes que utilizam este medicamento): reações anafilactoides Além destes, em relatos espontâneos, as seguintes reações adversas foram reportadas em casos isolados: nível de consciência reduzido, tontura, confusão, angioedema, inchaço de articulações, hiperidrose (suor excessivo), dor nas costas, bradicardia (queda de frequência cardíaca) e cromatúria (urina anormalmente corada) Informe ao seu médico, cirurgião-dentista ou farmacêutico o aparecimento de reações indesejáveis pelo uso do medicamento Informe também à empresa através do seu serviço de atendimento 9 O QUE FAZER SE ALGUÉM USAR UMA QUANTIDADE MAIOR DO QUE A INDICADA DESTE MEDICAMENTO Superdose pode ser tratada com medidas de suporte e, se requerido, um agente quelante para remoção do ferro No caso de superdose, podem ocorrer sintomas tais como náusea, vômito, diarreia, gastralgia (dor no estômago) e letargia Em casos graves, as seguintes situações podem ser esperadas: hiperglicemia (aumento dos níveis de glicose no sangue), leucocitose (diminuição de glóbulos brancos no sangue), acidose metabólica (acidez excessiva no sangue), hipotensão, taquicardia, convulsão, câimbra e coma De 12 a 48 horas após a administração, existe possibilidade da ocorrência de necrose tubular (lesão renal) e de células hepáticas (do fígado) O tratamento da superdose deve ser iniciado com a administração de deferoxamina se os seguintes sinais e/ ou sintomas ocorrerem dentro de 6 horas após a superdose: vômito, diarreia, glicemia > 150 mg/dL e leucocitose importante > 15 x 109/L; se o paciente não estiver em choque, devem ser administrados 1-2 g de deferoxamina, por via intramuscular, a cada 4 - 12 horas Se o paciente estiver em choque, uma dose inicial de 1 g de deferoxamina deve ser administrada por infusão intravenosa na velocidade máxima de infusão de 15 mg/kg de peso corporal, por hora Em ambos os casos, a dose máxima de deferoxamina deve ser de 6 g a cada 24 horas em adultos No caso de ocorrência de insuficiência renal, será necessária hemodiálise, uma vez que o complexo deferoxamina-ferro (ferrioxamina) é efetivamente eliminado pela diálise Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível Ligue para 0800 722 6001, se você precisar de mais orientações
Composição
COMPOSIÇÃO Cada mL de solução para diluição injetável contém sacarato de hidróxido férrico, equivalente a 20,0 mg Excipientes: hidróxido de sódio e água para injetáveis II) INFORMAÇÕES AO PACIENTE 1
Interação Medicamentosa
Sacarato de Hidróxido Férrico (substância ativa) não deve ser administrado concomitantemente a preparações orais de ferro, pois a absorção oral é reduzida.
Ação da Substância
Resultados de eficácia
Três estudos clínicos foram conduzidos para avaliar a eficácia e segurança de Sacarato de Hidróxido Férrico (substância ativa). Dois estudos foram realizados nos Estados Unidos (100 pacientes) e um na África do Sul (131 pacientes).
Estudo A
Estudo multicêntrico, aberto e controlado por histórico com 101 pacientes em hemodiálise (77 pacientes em tratamento com Sacarato de Hidróxido Férrico (substância ativa) e 24 no grupo controle histórico) com anemia ferropriva.
Os critérios de elegibilidade para tratamento incluíram
Hemodiálise crônica (vigente, 3 vezes por semana), recebendo eritropoietina, concentração de hemoglobina superior a 8,0 e inferior a 11,0 g/dL por pelo menos duas semanas consecutivas, saturação de transferrina < 20% e ferritina sérica < 300 ng/mL. A idade média dos pacientes no grupo de tratamento foi de 65 anos com faixa de 31 a 85 anos. A dose de eritropoietina foi mantida constante durante o estudo. O protocolo não requereu administração de uma dose teste, entretanto, alguns pacientes a receberam a critério do médico.
Os critérios de exclusão incluíram
Doença de base significativa, asma, doença inflamatória ativa ou infecção bacteriana ou viral. Sacarato de Hidróxido Férrico (substância ativa) 5 mL (uma ampola) contendo 100 mg de ferro elementar foi administrada através da linha de diálise em cada uma das sessões de diálise com uma dose cumulativa de 1000 mg de ferro elementar. Um máximo de 3 ampolas foi administrado por semana. Não foram permitidas preparações adicionais até após o 57o dia de avaliação. A alteração média de hemoglobina em relação ao basal no dia 24 (término do tratamento), dia 36 e dia 57 foi analisada.
A população de controle histórico consistiu de 24 pacientes com níveis de ferritina similares aos dos pacientes tratados, aos quais não foi realizada administração intravenosa de ferro por pelo menos 2 semanas e que haviam recebido terapia de eritropoietina com hematócrito variando entre 31 – 36 por pelo menos 2 meses antes da inclusão no estudo. A idade média dos pacientes no grupo controle histórico foi de 56 anos, com idades entre 29 a 80 anos. As idades e níveis de ferritina sérica foram similares entre os dois grupos. Dos 77 pacientes no grupo de tratamento, 44 (57%) eram homens e 33 (43%) mulheres. Os níveis basais médios de hemoglobina e de hematócrito foram maiores e a dose de eritropoietina foi menor na população de controle histórico que na população tratada com Sacarato de Hidróxido Férrico (substância ativa). Nesta, observou-se um maior aumento, estatisticamente significativo, em hemoglobina e hematócrito, em relação ao grupo de controle histórico. Vide Tabela 1.
Tabela 1. Alterações em relação ao basal (hemoglobina e hematócrito):
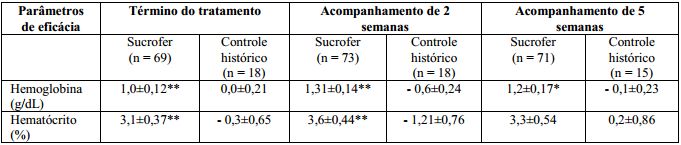 **p<0,01 e *p<0,05 comparado ao controle histórico a partir de análise ANCOVA com hemoglobina e ferritina sérica basais e dose de eritropoietina como co-variáveis.
**p<0,01 e *p<0,05 comparado ao controle histórico a partir de análise ANCOVA com hemoglobina e ferritina sérica basais e dose de eritropoietina como co-variáveis.
A ferritina sérica aumentou significativamente (p=0,0001) no desfecho do estudo a partir do basal na população tratada com Sacarato de Hidróxido Férrico (substância ativa) (165,3±24,2 ng/mL), comparada ao grupo controle (- 27,6±9,5 ng/mL). A saturação de transferrina também aumentou de forma significativa (p=0,0016) no desfecho do estudo a partir do basal na população tratada com Sacarato de Hidróxido Férrico (substância ativa) (8,8±1,6%), comparada ao grupo controle (- 5,1±4,3%).
Estudo B
Estudo multicêntrico, aberto, em 23 pacientes com deficiência de ferro e em hemodiálise, que haviam descontinuado o tratamento com ferro dextrana devido à intolerância. Os critérios de elegibilidade e a administração de Sacarato de Hidróxido Férrico (substância ativa) foram idênticos às do Estudo A. A idade média dos pacientes neste estudo foi de 53 anos, variando entre 21 e 79 anos. Dos 23 pacientes arrolados, 10 (44%) eram homens e 13 (56%) mulheres.
A divisão étnica dos pacientes incluídos no estudo foi a seguinte
- Caucasianos (8, 35%);
- Negros (8, 35%);
- Asiáticos (1, 4%);
- Hispânicos (6, 26%).
A alteração média a partir do basal ao final do tratamento (dia 24) nos parâmetros de hemoglobina, hematócrito e ferro sérico foi analisada.
Todos os 23 pacientes arrolados foram avaliados quanto à eficácia. Aumentos estatisticamente significativos na hemoglobina média (1,1±02, g/dL), hematócrito (3,6±0,6%), ferritina sérica (266,3±30,3 ng/mL) e a saturação de transferrina (8,7±2,0%) foram observados a partir do basal ao término do tratamento.
Estudo C
Estudo multicêntrico, aberto, de 2 períodos (tratamento seguido por um período de observação), em pacientes com deficiência de ferro e em hemodiálise.
Os critérios de elegibilidade para este estudo incluíram
Pacientes com hemodiálise crônica com hemoglobina menor ou igual a 10 g/dL, saturação de transferrina menor ou igual a 20% e ferritina sérica menor ou igual a 200 ng/mL, que estiveram em hemodiálise 2 a 3 vezes por semana. A idade média dos pacientes arrolados neste estudo foi 41 anos, variando entre 16 e 70 anos. Dos 130 pacientes avaliados quanto à eficácia neste estudo, 68 (52%) eram homens e 62 (48%) mulheres.
A divisão étnica dos pacientes incluídos foi a seguinte
- Caucasianos (30, 23%);
- Negros (30, 23%);
- Asiáticos (6, 5%);
- Etnias mistas (64, 49%).
Quarenta e oito por cento dos pacientes foram previamente tratados com ferro oral. Os critérios de exclusão foram similares àqueles dos Estudos A e B. Sacarato de Hidróxido Férrico (substância ativa) foi administrado em doses de 100 mg após as sessões de diálise, até que uma dose total pré-determinada (calculada) fosse administrada. Pacientes receberam Sacarato de Hidróxido Férrico (substância ativa) em cada sessão de diálise, duas a três vezes por semana.
Uma hora após o início de cada sessão, 5 mL de sacarose férrica (100 mg de ferro) em 100 mL de cloreto de sódio 0,9% foram administrados na linha de hemodiálise. Uma dose de 2,5 mL foi dada aos pacientes dentro de 2 semanas da inclusão no estudo. Pacientes foram tratados até que atingissem uma dose total de ferro individualmente calculada tendo como referência o nível basal de hemoglobina e peso corpóreo. Vinte e sete pacientes (20%) estiveram recebendo tratamento com eritropoietina no momento da entrada no estudo e estes continuaram recebendo a mesma dose de eritropoietina durante o estudo.
As alterações em relação ao basal nas semanas de observação 2 e 4 (término do estudo) foram analisadas. A população com intenção de tratar, modificada, consistiu de 131 pacientes. Aumentos significativos (p<0,0001) a partir do basal na hemoglobina média (1,7 g/dL), hematócrito (5%), ferritina sérica (434,6 ng/mL) e saturação de transferrina sérica (14%) foram verificados na semana 2 do período de observação e estes valores permaneceram significativamente elevados (p<0,0001) na semana 4 do período de observação.
Características farmacológicas
O ferro presente em Sacarato de Hidróxido Férrico (substância ativa) está na forma trivalente como um complexo coloidal macromolecular de sacarato de hidróxido de ferro III. O núcleo do hidróxido de ferro III polinuclear é superficialmente rodeado por um grande número de moléculas de sacarose ligadas não covalentemente, resultando em um complexo cuja massa molecular é aproximadamente 43 kDa, suficientemente grande para inibir a sua eliminação renal. O complexo resultante é estável e não libera íons de ferro sob condições fisiológicas. O ferro nos núcleos polinucleares está ligado a uma estrutura similar como ocorre fisiologicamente com a ferritina.
O ferro trivalente do complexo coloidal de sacarato de hidróxido de ferro III, presente no Sacarato de Hidróxido Férrico (substância ativa), combina-se, sem alteração de valência, com a transferrina. Parte dele forma ferro de depósito (ferritina) e outra parte destina-se à gênese da hemoglobina, de mioglobina e de enzimas contendo ferro. A aplicação pela via intravenosa promove utilização instantânea do ferro, o que constitui um fator relevante, particularmente em casos de anemias muito pronunciadas.
O ligante do complexo é a sacarose (dissacarídeo), não contendo nenhum dextrano (polissacarídeo), portanto, não ocorre nenhuma reação com o anticorpo específico para dextrano, que determinaria uma reação anafilática induzida pelo mesmo.
Propriedades Farmacodinâmicas
Após a administração intravenosa de Sacarato de Hidróxido Férrico (substância ativa), a sacarose férrica é dissociada pelo sistema retículo- endotelial em ferro e sacarose. Em 22 pacientes em hemodiálise, recebendo eritropoietina (recombinante humana), tratados com sacarose férrica (equivalente a 100 mg de ferro) três vezes/semana por três semanas, aumentos significativos no ferro e ferritina séricos e diminuição importante na capacidade total de ligação a ferro ocorreu após quatro semanas do início do tratamento.
Propriedades Farmacocinéticas
Em adultos saudáveis tratados com doses intravenosas de Sacarato de Hidróxido Férrico (substância ativa), seu componente ferro exibe cinética de primeira ordem com uma meia-vida de eliminação de 6 horas, clearance total de 1,2 L/h, volume aparente de distribuição no estado não-estacionário de 10,0 L e volume aparente de distribuição no estado estacionário de 7,9 L.
Uma vez que a eliminação de ferro do soro depende da necessidade de ferro nos estoques e da sua utilização pelos tecidos, se espera que o clearance sérico de ferro seja mais rápido em pacientes com deficiência de ferro em comparação aos indivíduos saudáveis. Os efeitos de idade e gênero na farmacocinética de Sacarato de Hidróxido Férrico (substância ativa) não foram estudados.
Sacarato de Hidróxido Férrico (substância ativa) não é dialisável por membranas de diálise de alto fluxo, por exemplo, CA201 High Efficiency (Baxter) ou F80A (Fresenius). Em estudos in vitro, a quantidade de sacarose férrica no fluido dialisado foi inferior aos níveis de detecção no ensaio (menor que 2 partes por milhão).
Distribuição
Em adultos saudáveis recebendo doses intravenosas de Sacarato de Hidróxido Férrico (substância ativa), seu componente ferro parece se distribuir principalmente no sangue e, em alguma extensão, no fluido extravascular. Um estudo avaliando Sacarato de Hidróxido Férrico (substância ativa) contendo 100 mg de ferro marcado 52Fe/59Fe em pacientes com deficiência de ferro demonstra que uma quantidade significativa do ferro administrado se distribui no fígado, baço e medula óssea e que esta é um compartimento que captura o fero e não um volume de distribuição reversível.
Metabolismo e eliminação
O componente sacarose é eliminado principalmente por excreção renal. Em um estudo avaliando uma dose intravenosa única de Sacarato de Hidróxido Férrico (substância ativa) contendo 1,510 mg de sacarose e 100 mg de ferro em 12 adultos saudáveis (9 mulheres, 3 homens) com idades entre 32 e 52 anos, 68,3% da sacarose foi eliminada na urina em 4 horas e 75,4% em 24 horas. Parte do ferro também é eliminada na urina. Os níveis de transferrina e do seu receptor não se alteraram imediatamente após a administração da dose. Neste e em outro estudo analisando uma única dose intravenosa de sacarose férrica, contendo 500 – 700 mg de ferro em 26 pacientes com anemia em terapia com eritropoietina (23 mulheres, 3 homens; faixa de idade 16 – 60), aproximadamente 5% do ferro foi eliminado na urina em 24 horas em cada intervalo de dose.
Dados de segurança pré-clínica
Carcinogenicidade, mutagenicidade e prejuízo à fertilidade
Não foram realizados estudos de longo prazo em animais para avaliar o potencial carcinogênico de Sacarato de Hidróxido Férrico (substância ativa).
Testes in vitro (Ames, mutação em célula de linfoma de camundongo (L5178Y/K±), aberração cromossômica em linfócito humano ou micronúcleos em camundongos) não mostraram potencial genotóxico.
Sacarato de Hidróxido Férrico (substância ativa) em doses intravenosas de até 15 mg ferro/kg/dia, cerca de 1,2 vezes a dose máxima recomendada em humanos com base na superfície corpórea, não apresentou efeitos na fertilidade ou capacidade reprodutiva de ratos machos e fêmeas.
Cuidados de Armazenamento
ONDE, COMO E POR QUANTO TEMPO POSSO GUARDAR ESTE MEDICAMENTO SUCROFER deve ser conservado em temperatura ambiente (15ºC a 30ºC), protegido da luz e da umidade Não congelar Número de lote e datas de fabricação e validade: vide embalagem Não use medicamento com o prazo de validade vencido Guarde-o em sua embalagem original SUCROFER é uma solução viscosa, de cor marrom escura e livre de partículas estranhas visíveis Devido a características químicas de SUCROFER é normal que ocorra a aglomeração do ferro, que desaparece com agitação SUCROFER deve ser administrado apenas em ambiente hospitalar Antes de usar, observe o aspecto do medicamento Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo Todo medicamento deve ser mantido fora do alcance das crianças 6
Dizeres Legais
III) DIZERES LEGAIS MS nº 1 4277 0035 Farmacêutica Responsável: Lívia Grégio Honma - CRF-SP nº 40 863 Fabricado por: Claris Injectables Limited - Unidade 1 Vasana-Chacharwadi, Ahmedabad-382 213, Índia 5 Registrado e Importado por: Claris Produtos Farmacêuticos do Brasil Ltda Alameda Araguaia, 3852 – Tamboré CEP: 06455-000 – Barueri - SP CNPJ: 02 455 073/0001-01 Comercializado por: Meizler-UCB Biopharma S A Alameda Araguaia, 3833 – Tamboré – Barueri - SP VENDA SOB PRESCRIÇÃO MÉDICA USO RESTRITO A HOSPITAIS 0302017029 R2 Maio 2015 Histórico de alteração para bula Dados da submissão eletrônica Dados da petição/notificação que altera bula Dados das alterações de bulas Data do expediente No expediente Assunto Data do expediente N° do expediente Assunto Data de aprovação Itens de bula Versões (VP/VPS)
Dizeres legais VP e VPS 20 MG/ML SOL INJ CT 5 AMP VD INC X 5 ML Petição atual Petição atual 10454 - ESPECÍFICO - Notificação de Alteração de Texto de Bula – RDC 60/12 - - - - 5 Onde, como e por quanto tempo posso guardar este medicamento 7 Cuidados de armazenamento do medicamento 8 Posologia e Modo de Usar VP e VPS 20 MG/ML SOL INJ CT 5 AMP VD INC X 5 ML